Нові публікації
Нові дані сприяють кращому розумінню причин синдрому Ретта
Останній перегляд: 02.07.2025

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.

Синдром Ретта – це рідкісне нейророзвиточне захворювання, від якого наразі немає ліків або належного лікування. Він викликає серйозні фізичні та когнітивні симптоми, багато з яких перетинаються з розладами аутистичного спектру.
Синдром Ретта спричинений мутаціями в гені MECP2, який високо експресується в мозку та, ймовірно, відіграє важливу роль у підтримці здоров'я нейронів. Ген розташований на Х-хромосомі, і синдром вражає переважно дівчат. Щоб розробити методи лікування синдрому Ретта, дослідники хочуть краще зрозуміти MECP2 та його функції в мозку.
Дослідники, включаючи співзасновника Інституту Вайтхеда Рудольфа Єніша, вивчають MECP2 протягом десятиліть, проте багато основних фактів про ген залишаються невідомими. Білок, кодований геном, MECP2, бере участь у регуляції генів; він зв'язується з ДНК і впливає на рівні експресії різних інших генів або на кількість білка, який вони виробляють.
Однак дослідники не мали повного списку генів, на які впливає MECP2, і не було єдиної думки щодо того, як MECP2 впливає на ці гени.
Ранні дослідження MECP2 вказували на те, що він є репресором, знижуючи експресію своїх генів-мішеней, але дослідження Яніша та інших раніше показали, що MECP2 також діє як активатор, збільшуючи експресію своїх цілей, — і що він може бути активатором взагалі. Також невідомим був механізм дії MECP2 або що саме робить білок, щоб викликати зміни в експресії генів.
Обмеження технологій завадили дослідникам отримати ясність у цих питаннях. Але Яніш, постдок його лабораторії І Лю та колишній член лабораторії Яніша Ентоні Флам'є, який зараз є доцентом дослідницького центру CHU Sainte-Justine в Університеті Монреаля, використали передові методи, щоб відповісти на ці питання, що залишилися, щодо MECP2 та отримати нове розуміння його ролі у здоров'ї та захворюваннях мозку.
Їхні результати були опубліковані в журналі Neuron, а дослідники також створили онлайн-репозиторій своїх даних MECP2, портал MECP2-NeuroAtlas, як ресурс для інших дослідників.
«Я думаю, що ця стаття докорінно змінить розуміння людьми того, як MECP2 викликає синдром Ретта. Ми маємо абсолютно нове розуміння механізму, і це може відкрити нові шляхи для розробки методів лікування цього захворювання», — каже Яніш, який також є професором біології в Массачусетському технологічному інституті.
Глибше розуміння MECP2 у мозку
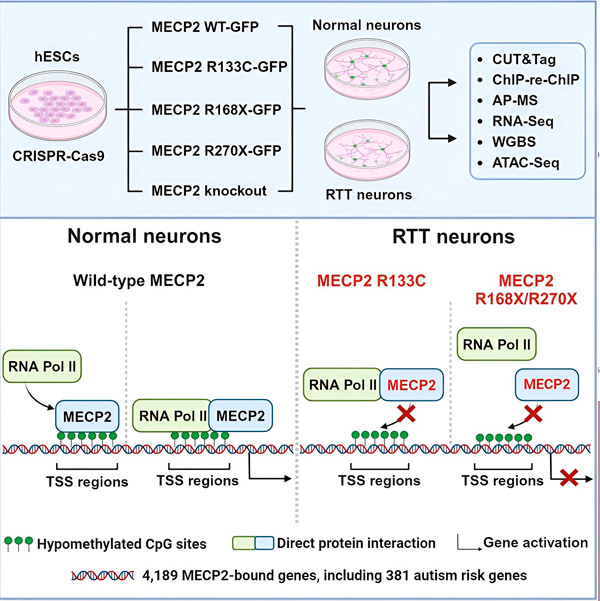
Дослідники спочатку створили детальну карту місць зв'язування MECP2 у послідовностях генів нейронів людини, або всередині генів, або в регуляторних ділянках ДНК поблизу них. Вони використали підхід під назвою CUT&Tag, який може з високою точністю визначати взаємодії білків з ДНК.
Дослідники виявили понад 4000 генів, пов'язаних з MECP2. Вони повторили їхнє картування в нейронах із поширеними мутаціями MECP2, пов'язаними із синдромом Ретта, щоб визначити, де MECP2 виснажується під час захворювання.
Знання того, з якими генами зв'язується MECP2, дозволило Лю та Флам'є почати встановлювати зв'язки між мішенями MECP2 та здоров'ям мозку. Вони виявили, що багато з його мішеней беруть участь у розвитку та функціонуванні нейрональних аксонів і синапсів.
Вони також порівняли свій список мішеней MECP2 з базою даних Ініціативи з дослідження аутизму Фонду Саймонса (SFARI) генів, пов'язаних з аутизмом, і виявили, що 381 ген у цій базі даних є мішенями MECP2.
Джерело: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Ці висновки можуть допомогти з'ясувати механізми, що лежать в основі симптомів аутизму при синдромі Ретта, та стати гарною відправною точкою для дослідження можливої ролі MECP2 в аутизмі.
«Ми створили першу інтегровану карту епігеному MECP2 у здоров’ї та хворобах, і ця карта може бути орієнтиром для майбутніх досліджень», — каже Лю. «Знання того, які гени є мішенями MECP2, а які гени безпосередньо порушуються при захворюванні, забезпечує міцну основу для розуміння синдрому Ретта та постановки питань про регуляцію генів у нейронах».
Дослідники також досліджували, чи збільшує, чи зменшує MECP2 експресію своїх цільових генів. Відповідно до того, як деякі люди ідентифікували MECP2 як активатор, а інші — як репресор, Лю та Флам'є знайшли приклади, коли MECP2 відігравав обидві ролі.
Однак, хоча MECP2 частіше вважають репресором, Лю та Флам'є виявили, що він здебільшого є активатором, що підтверджує попередні висновки Яніша та Лю. Один новий експеримент показав, що MECP2 активує щонайменше 80% своїх мішеней, а інший виявив, що він активує до 88% своїх мішеней.
Карта генів-мішеней, створена дослідниками, дала додаткове розуміння ролі MECP2 як активатора. Вони виявили, що гени, які активує MECP2, зазвичай зв'язуються з ділянкою ДНК вище за течією від гена, яка називається сайтом початку транскрипції.
Це місце, де клітинний механізм ініціює процес транскрипції гена в РНК, після чого РНК транслюється у функціональний білок, який є продуктом експресії генів. Присутність MECP2 у місці початку транскрипції, де починається експресія генів, узгоджується з його роллю як активатора генів.
Потім дослідники вирішили визначити, яку роль MECP2 відіграє в активації генів. Вони дослідили, з якими молекулами, окрім ДНК, зв'язується MECP2 у цьому місці, і виявили, що MECP2 безпосередньо взаємодіє з білковим комплексом під назвою РНК-полімераза II (РНК Pol II). РНК Pol II є ключовою клітинною машиною, яка транскрибує ДНК у РНК. РНК Pol II не може самостійно знаходити гени, тому їй потрібен різноманітний кофактор, або білковий партнер, щоб допомогти їй виконувати свою роботу.
Дослідники припускають, що MECP2 служить одним із таких кофакторів, допомагаючи РНК-полімеразу II ініціювати транскрипцію в генах, де зв'язується MECP2. Структурний аналіз MECP2 виявив частини молекули, які зв'язуються з РНК-полімеразу II, а інші експерименти підтвердили, що втрата MECP2 знижує присутність РНК-полімерази II у відповідних сайтах початку транскрипції, а також рівні експресії цільових генів.
Це говорить про те, що синдром Ретта може бути спричинений зниженням транскрипції генів, на які впливає MECP2, через мутації MECP2, які запобігають його зв'язуванню з РНК-полімеразу II або зв'язуванню з ДНК. Відповідно до цієї ідеї, найпоширенішими мутаціями MECP2, пов'язаними із захворюванням, є усічення: мутації, в яких відсутня частина білка, що може змінити взаємодію між MECP2 та РНК-полімеразу II.
Дослідники сподіваються, що їхні висновки не лише змінять наше розуміння MECP2, але й що глибше та ширше розуміння того, як MECP2 впливає на розвиток та функціонування мозку, може призвести до нових знань, які допоможуть людям із синдромом Ретта та пов'язаними з ним розладами, включаючи аутизм.
«Цей проект є чудовим прикладом спільної роботи лабораторії Яніша», — каже Флам’єр. «У нас з Рудольфом була специфічна проблема, пов’язана із синдромом Ретта, і я мав досвід роботи з технологією CUT&Tag, яка могла вирішити цю проблему. Завдяки обговоренню ми зрозуміли, що можемо об’єднати наші зусилля, і тепер у нас є чудове сховище інформації про MECP2 та його зв’язок із захворюваннями».