Нові публікації
Виявлено нове генетичне пояснення серцевого захворювання
Останній перегляд: 02.07.2025

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
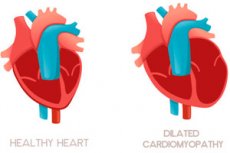
Нове дослідження, опубліковане в журналі Nature Genetics під керівництвом вчених з Університетського коледжу Лондона (UCL), Імперського коледжу Лондона та Лабораторії медичних наук MRC, виявило, що дилатаційна кардіоміопатія (ДКМП) може бути спричинена комбінованим впливом сотень або тисяч генів, а не однією «несправною» генетичною мутацією, як вважалося раніше.
Що таке дилатаційна кардіоміопатія?
ДКМП – це стан, при якому серце збільшується та слабшає, знижуючи його здатність ефективно перекачувати кров. У Великій Британії це захворювання вражає до 260 000 людей (1 з 250) і є основною причиною трансплантації серця.
Раніше вважалося, що ДКМП спричинена дефектними копіями одного гена, які передаються у спадок. Однак більше половини пацієнтів не мають цієї генетичної мутації.
Ключові висновки дослідження
Кумулятивний вплив генів:
- Приблизно від чверті до третини ризику розвитку ДКМП можна пояснити незначними ефектами багатьох генетичних відмінностей, розподілених по всьому геному.
- Це вказує на схожість між ДКМП та більш поширеними захворюваннями, такими як ішемічна хвороба серця, де кілька генів разом впливають на ризик.
Полігенний ризик:
- Вчені розробили полігенну шкалу ризику для оцінки ймовірності розвитку ДКМП на основі комбінованого впливу багатьох генів.
- Люди з найвищим полігенним ризиком (верхній 1%) мають у чотири рази вищий ризик розвитку ДКМП, ніж люди із середнім ризиком.
Ризик за наявності мутацій:
- У пацієнтів з рідкісними мутаціями ризик розвитку захворювання зростає до 7,3% (порівняно з 1,7% у пацієнтів з низьким полігенним ризиком).
Відкриття нових генів:
- Було ідентифіковано вісімдесят геномних регіонів та 62 гени, пов'язані з DCM, більшість з яких раніше не були описані.
Практичне застосування результатів
Покращена діагностика:
- Полігенні шкали ризику можуть допомогти лікарям точніше прогнозувати ризик захворювання у пацієнтів та їхніх сімей.
- Це особливо корисно для тих, чиє захворювання спричинене комбінованим впливом генів, а не однією мутацією.
Визначення груп високого ризику:
- Пацієнти з високим полігенним ризиком можуть перебувати під ретельнішим наглядом та мати доступ до клінічних випробувань профілактичного лікування.
Потенційні нові препарати:
- Вивчення генів, пов'язаних з DCM, може пролити світло на біологічні процеси, що лежать в основі захворювання, та допомогти розробити нові методи лікування.
Коментарі дослідників
Д-р Том Ламбурс (UCL):
«Наші висновки змінюють розуміння генетики диклофіброзної миєломи (ДКМП): захворювання спричинене не однією мутацією, а взаємодією багатьох генів. Це відкриває нові можливості для прогнозування та профілактики ризиків».Професор Джеймс Вейр (Імперський коледж Лондона):
«Ми сподіваємося, що наші відкриття покращать точність генетичного тестування та збільшать кількість пацієнтів, яким можна буде дати генетичне пояснення їхнього захворювання».Професор Метін Авкіран (Британський фонд серця):
«Ці результати дають чіткіше уявлення про індивідуальний ризик для пацієнтів і можуть стати основою для персоналізованого моніторингу та лікування ДКМП».
Майбутні напрямки досліджень
- Інтеграція полігенних оцінок ризику в генетичне тестування.
- Подальше вивчення ролі нещодавно виявлених генів у розвитку ДКМП.
- Розробка персоналізованих методів лікування та моніторингу.
Дослідження є важливим кроком уперед у розумінні ДКМП, прокладаючи шлях для точнішої діагностики та розробки нових методів лікування для тисяч пацієнтів.