Нові публікації
Виявлено ключовий білок, що запобігає втраті кісткової маси при остеопорозі
Останній перегляд: 02.07.2025

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
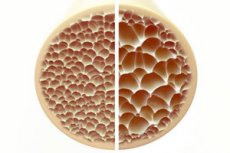
Остеопороз, стан, що характеризується пористими та крихкими кістками, становить значну загрозу для здоров'я скелета. Кістки, як основна структурна опора людського тіла, забезпечують життєво важливу підтримку. Коли кісткова маса зменшується, це не тільки погіршує цю підтримку, але й погіршує загальну функцію, що призводить до зниження якості життя.
Зі зростанням захворюваності на остеопороз серед населення похилого віку, навантаження на ресурси охорони здоров'я, пов'язані з довгостроковим доглядом, зростає. Тому необхідно розуміти механізми, що сприяють розвитку остеопорозу, та розробляти ефективні цілеспрямовані методи лікування для мінімізації його довгострокового впливу.
Остеобласти та остеокласти – це два типи клітин, які відіграють ключову роль у підтримці та ремоделюванні кісткової тканини. У той час як остеобласти – це клітини, що утворюють кістку, відповідальні за синтез та відкладення нової кісткової тканини, остеокласти – це клітини, що руйнують кістку, що беруть участь у розщепленні та видаленні старої або пошкодженої кісткової тканини.
Збільшення частки остеокластів призводить до втрати кісткової маси при таких станах, як остеопороз, ревматоїдний артрит (запалення суглобів) та метастази в кістках (рак, що поширився на кістки). Остеокласти виникають внаслідок диференціації макрофагів або моноцитів, які є типами імунних клітин.
Таким чином, пригнічення диференціації остеокластів може служити терапевтичною стратегією для запобігання втраті кісткової маси. Однак точні молекулярні механізми, що регулюють складний процес ремоделювання кісток, залишаються неясними.
У новому дослідженні професор Тадайоші Хаята, пан Такуто Конно та пані Хітомі Мурачі з Токійського університету наук разом з колегами заглибилися в молекулярну регуляцію диференціації остеокластів. Стимуляція за допомогою ліганду рецептора-активатора ядерного фактора каппа B (RANKL) індукує диференціацію макрофагів в остеокласти.
Крім того, сигнальні шляхи кісткового морфогенетичного білка (BMP) та трансформуючого фактора росту (TGF)-β були задіяні в регуляції диференціації остеокластів, опосередкованої RANKL. У цьому дослідженні дослідники мали на меті дослідити роль Ctdnep1, фосфатази (ферменту, що видаляє фосфатні групи), яка, як повідомлялося, пригнічує сигнальні шляхи BMP та TGF-β.
Дослідження опубліковане в журналі Biochemical and Biophysical Research Communications.
Професор Хаята стверджує: «RANKL діє як «прискорювач» диференціації остеокластів. Керування автомобілем вимагає не лише акселератора, а й гальм. Тут ми виявили, що Ctdnep1 діє як «гальмо» в диференціації остеокластів».
Дослідники спочатку дослідили експресію Ctdnep1 у макрофагах мишей, оброблених RANKL, та контрольних клітинах, що не були оброблені. Вони спостерігали, що експресія Ctdnep1 не змінювалася у відповідь на стимуляцію RANKL. Однак, він був локалізований у цитоплазмі у гранулярній формі в макрофагах та диференційований в остеокласти, що відрізняється від його нормальної перинуклеарної локалізації в інших типах клітин, що вказує на його цитоплазматичну функцію в диференціації остеокластів.
Крім того, нокдаун Ctdnep1 (зниження експресії генів) призвів до збільшення кількості остеокластів, позитивних на тартрат-резистентну кислу фосфатазу (TRAP), де TRAP є маркером диференційованих остеокластів.
Нокаут Ctdnep1 призвів до збільшення експресії ключових маркерів диференціації, включаючи "Nfatc1", головного транскрипційного фактора, індукованого RANKL для диференціації остеокластів. Ці результати підтверджують "гальмівну функцію" Ctdnep1, завдяки якій він негативно регулює диференціацію остеокластів. Більше того, нокаут Ctdnep1 також призвів до збільшення абсорбції фосфату кальцію, що свідчить про супресивну роль Ctdnep1 у резорбції кісток.
Зрештою, хоча нокаут Ctdnep1 не змінив сигналізацію BMP та TGF-β, клітини з дефіцитом Ctdnep1 демонстрували підвищений рівень фосфорильованих (активованих) білків, які є продуктами сигнального шляху RANKL. Ці результати свідчать про те, що інгібуючий ефект Ctdnep1 на диференціацію остеокластів може бути опосередкований не через сигналізацію BMP та TGF-β, а через зниження регуляції сигнального шляху RANKL та рівнів білка Nfatc1.
Загалом, ці результати дають нове розуміння процесу диференціації остеокластів та визначають потенційні терапевтичні мішені, які можна було б використовувати для розробки методів лікування, спрямованих на зменшення втрати кісткової маси, спричиненої гіперактивністю остеокластів. Окрім захворювань, що характеризуються втратою кісткової маси, Ctdnep1 також був ідентифікований як причинний фактор медулобластоми, пухлини головного мозку у дітей. Автори з оптимізмом сподіваються, що їхні дослідження можна буде поширити на інші захворювання людини, окрім метаболізму кісток.
Професор Хаята робить висновок: «Наші результати свідчать про те, що Ctdnep1 необхідний для запобігання надмірному остеокластогенезу. Ці результати можуть ще більше розширити наші знання про те, як мережа фосфорилювання-дефосфорилювання контролює диференціацію остеокластів, і можуть запропонувати нові терапевтичні стратегії для лікування захворювань кісток, пов’язаних з надмірною активністю остеокластів».