Медичний експерт статті

Нові публікації
Арахнодактилія
Останній перегляд: 04.07.2025

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
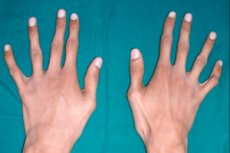
Однією з рідкісних спадкових патологій сполучної тканини є арахнодактилія – деформація пальців, що супроводжується видовженням трубчастих кісток, викривленнями скелета, порушеннями роботи серцево-судинної системи та органів зору.
Вперше захворювання було описано доктором Вільямсом та французьким педіатром Марфаном, які назвали його доліхостеномелією. Пізніше у світовій медицині з'явився термін синдром Марфана, а з 1902 року використовується назва арахнодактилія. Патологія пов'язана з вродженими порушеннями сполучної тканини, і однією з провідних ознак вважаються видовжені, стоншені та викривлені «павукові» пальці. [ 1 ]
Епідеміологія
Арахнодактилія вважається спадковою моногенною патологією сполучної тканини з аутосомно-домінантним типом успадкування, високою пенетрантністю та різним ступенем експресивності.
Приблизно у 75-80% випадків захворювання є спадковим, а в решті випадків виникає в результаті спонтанних мутацій (зокрема, внаслідок точкових міссенс-мутацій). [ 2 ]
Доведено, що патогенетичні особливості арахнодактилії зумовлені змінами в кількох генах (у 95% випадків):
- у гені фібриліну-1 (вивчено близько тисячі мутацій) на хромосомі 15q21.1;
- ген TGFβR1 або TGFβR2 на хромосомі 9 та 3p24.2-P25.
Ці зміни безпосередньо впливають на ступінь клінічних проявів.
Мутації в α-2-ланцюзі колагену I типу були описані у 5% пацієнтів.
Поширеність арахнодактилії становить приблизно 2 випадки на 5 тисяч осіб. Расової чи гендерної детермінації немає. [ 3 ]
Причини арахнодактилії
Арахнодактилія виникає в результаті спонтанної мутації. Захворювання не характеризується загальновідомими ознаками вроджених спадкових патологій, а видовжені пальці проявляються переважно при синдромі Марфана та гомоцистинурії.
Це захворювання є рідкісним, і всі виявлені пацієнти мали мутацію гена FBN1 (ген фібриліну), розташованого на хромосомі 15 в області q21.1. Клінічний поліморфізм синдрому можна пояснити масою мутацій, причому близько 15% – це мутаційні зміни, що виникли під час зачаття. [ 4 ]
Атипові різновиди арахнодактилії зумовлені мутаціями в інших генах: виявлено зв'язок між розвитком патології та мутаціями в гені LTBP3, R, який локалізується на 14-й хромосомі в області q24 та трансформує фактор росту.
Переважна більшість мутацій – це так звані місенс-мутації (приблизно 57%). Понад 20% – це невеликі делеції з втратою хромосомних частин, близько 12% – це мутації сайту сплайсингу, 8% – це нонсенс-мутації, а 2% – це масштабні перебудови та делеції. [ 5 ]
Вроджена контрактуральна арахнодактилія спричинена порушенням синтезу білка фібриліну-2. Проблема спричинена мутацією в гені FBN2, розташованому на п'ятій хромосомі в ділянці q23-q31. Усі мутації є переважно точковими, що змінюють кодон на код іншої амінокислоти. [ 6 ]
Гомоцистинурія розвивається на тлі порушення метаболізму незамінної амінокислоти метіоніну. Основою патології може бути втрата активності або її зниження наступних ферментних речовин:
- Фермент цистатіонін-бета-синтази (ЦБС). Патологія є B6 резистентною та проявляється ознаками, характерними для B6 незалежної форми, проте введення препарату вітаміну B6 не призводить до покращення.
- Фермент N5, N10 метилентетрагідрофолатредуктаза (MTHFR). Цей ферментативний агент є субстратом, що викликає деметилювання гомоцистеїну та його перетворення на метіонін, незамінну амінокислоту, що входить до складу багатьох білкових та пептидних речовин.
- Фермент N5 метилентетрагідрофолат. Йдеться про B6 залежну форму патології, яка розвивається при дефіциті вітаміну B6 . Введення вітамінного препарату в організм дозволяє покращити стан пацієнта.
- Фермент гомоцистеїнтрансметилази. Розвивається на тлі порушень обміну кобаламіну – вітаміну B12.
Фактори ризику
Арахнодактилія – генетично обумовлене захворювання, що характеризується системним ураженням сполучної тканини. Патологія пов'язана зі змінами гена фібриліну 1, локалізованого на короткому плечі 15-ї хромосоми в локусі 21.1.
Арахнодактилія успадковується аутосомно-домінантним типом, з високою пенетрантністю та варіабельною експресивністю. Патологія може проявлятися як з чоловічої, так і з жіночої сторони.
Патогенез
Більше 50% маси тіла людини складають сполучнотканинні структури, зокрема скелет, шкіра, судинна мережа, лімфа та кров.
Клітини сполучної тканини представлені фібробластами та їх підтипами: остеобластами, хондроцитами, кератобластами, одонтобластами, а також макрофагами та тучними клітинами.
Ембріональна сполучна тканина є матеріалом для формування конституційних, генетичних та епігенетичних компонентів. Захворювання сполучної тканини тією чи іншою мірою відображаються на всьому організмі в цілому, на його функціональній спроможності та конституції.
При арахнодактилії відбувається заміщення нуклеотидів у гені, що містить структурну інформацію про пептид фібрилін-1. Ця білкова речовина належить до групи глікопротеїнів, бере участь у мікрофібрилярній системі та утворює основу еластичних фібрил сполучної тканини.
Тканина підтримує стабільну структуру завдяки міжклітинному матриксу, який містить багато факторів росту, що забезпечують систематичне оновлення клітин. Великі судини та зв'язки містять численні еластинові фібрили, пошкодження яких викликає основні симптоми арахнодактилії.
Трансформуючий фактор росту β значно пошкоджений: його неактивна форма не зв'язана, що призводить до підвищення біологічної активності цього фактора.
Фібрилярні порушення призводять до аномального утворення волокон, що призводить до втрати міцності та еластичності шкіри та інших структур сполучної тканини.
Коли порушується структура колагену, порушується і основна ланка гемостазу.
Фахівці відзначають причетність гормонального дисбалансу до виникнення та прогресування аномалій сполучнотканинних структур, зокрема арахнодактилії. Тромботичні симптоми визначаються реологічними порушеннями – зокрема, змінами в'язкості крові в змінених судинах брахіоцефальної області.
Травний тракт уражається через колагенові порушення: спостерігаються гіпомоторна дискінезія жовчовивідних шляхів, грижі стравохідного отвору діафрагми, гепатобіліарні аномалії, хронічна форма «стертого» гастродуоденіту, доліхосигма.
Як успадковується арахнодактилія?
Арахнодактилія успадковується за аутосомно-домінантним типом, але може виникати і в результаті спонтанних мутацій. Тобто патологія не обов'язково має проявлятися у когось із близьких родичів – наприклад, у батьків, бабусь і дідусів. Мутації, що лежать в основі патології, можуть дати про себе знати випадковим чином.
Симптоми арахнодактилії
У медицині розрізняють латентну та явну арахнодактилію. При латентній арахнодактилії зміни присутні, але вони ізольовані та обмежені однією або двома системами органів. Явна арахнодактилія проявляється множинними видимими порушеннями з різною інтенсивністю симптомів. У цьому випадку стан пацієнта може бути відносно стабільним протягом десятиліть або неухильно прогресувати, проявляючись щодо інших органів і систем.
Часто синдром арахнодактилії можна виявити протягом перших кількох днів після народження дитини.
Головною зовнішньою ознакою арахнодактилії є тонкі та видовжені пальці, які через короткі сухожилля набувають типової кривизни та нагадують павукові лапки.
Павутинні пальці або арахнодактилія стають чітко помітними майже з періоду новонародженості. Але ознака стає більш очевидною приблизно у трирічному віці.
Характерні також показники зросту: діти зазвичай високого зросту, передпліччя та гомілки довгі, кінцівки непропорційні та худі. Диспропорція особливо очевидна при високій локалізації талії та колінних чашечок. [ 7 ]
Інші захворювання скелета включають:
- звужена та видовжена форма черепа (доліхоцефалічна) з аномально сформованою лицьовою ділянкою;
- лійкоподібна конфігурація грудної клітки (так звана «птах»);
- викривлений хребет;
- вроджені вивихи стегна;
- звичні колінно-плечові вивихи;
- викривлення п'яткової кістки;
- остеофітні утворення – кісткові розростання;
- плоскостопість;
- недостатній розвиток жирового прошарку.
Додаткові ознаки арахнодактилії можуть включати:
- короткозорість;
- синюваті склери;
- сублюксація кришталика;
- порушення роботи серця та судин (вади серця, аневризма аорти);
- виражена худорлявість;
- гіпермобільність суглобів;
- «арочне» небо.
Арахнодактилія характеризується видовженими трубчастими кістками, що спричиняють різні викривлення скелета. Крім того, у багатьох пацієнтів на тлі нормального психічного розвитку спостерігаються пірамідні порушення, асиметрія сухожильних рефлексів, анозокорія та ністагм.
Якщо мова йде про арахнодактилію, що супроводжує гомоцистинурію, то у пацієнта можуть спостерігатися такі симптоми:
- періодичні напади судом;
- вторинна глаукома з сублюксацією кришталика;
- відшарування сітківки, астигматизм;
- пошкодження артеріальних стовбурів (включаючи нирки, головний мозок);
- остеопороз;
- тромбоз;
- розумова відсталість.
Арахнодактилія у новонароджених
У більшості випадків арахнодактилію можна виявити у дитини протягом перших кількох днів її життя. Однак з віком симптоми, як правило, погіршуються.
Перші ознаки, які дозволяють запідозрити захворювання, зазвичай такі:
- надзвичайно довгі кінцівки та пальці;
- недостатня вага за умови належного фізичного стану дитини;
- видовжене, довге обличчя;
- брак жиру в організмі, погано розвинені м'язи;
- надмірна гнучкість суглобів.
Приблизно з чотирьох років на тлі арахнодактилії починає змінюватися грудна клітка, викривляється хребетний стовп, розвивається плоскостопість.
З боку органів зору спостерігаються міопія, ектопія кришталика, змінена конфігурація рогівки, косоокість, гіпопластичні процеси в райдужній оболонці та сітківці. Такі порушення можна помітити вже в перші 2-3 роки життя дитини, хоча з роками вони прогресують.
Найбільшу небезпеку становлять порушення серцево-судинної системи. Якщо такі порушення присутні, то за відсутності відповідного лікування існують високі ризики смерті пацієнта вже в дитинстві. Серед особливо небезпечних патологій можна виділити пошкодження судинних стінок, вади розвитку серця, дефекти коронарних судин. Іноді у дітей першого року життя вже виявляється наростаюча серцева недостатність.
Арахнодактилія може супроводжуватися ураженням інших органів, зокрема, нервової системи, бронхів і легень, шкіри та сечостатевої системи. Такі порушення виявляються під час діагностичних процедур.
Ускладнення і наслідки
Найпоширенішими побічними наслідками арахнодактилії є ураження серця та органів дихання. Крім того, з'являються проблеми із зором, аж до його втрати.
Пацієнти з арахнодактилією потребують комплексної терапії всіх існуючих патологій. Якщо таке лікування не проводиться, тривалість життя пацієнтів значно скорочується: рідкісним пацієнтам без медичного втручання вдається дожити до 40 років. З роками проблеми з серцево-судинною системою погіршуються, зростає ризик великих крововиливів з трагічним результатом.
Люди, які страждають на арахнодактилію, повинні регулярно проходити обстеження та лікувати супутні патології: лише за цієї умови у них є всі шанси прожити довге життя без серйозних ускладнень.
Синдром Марфана та арахнодактилія потребують особливої уваги, якщо пацієнтка вагітніє. У такій ситуації важливо регулярно та ретельно обстежувати пацієнтку, а також медикаментозно підтримувати стан її серцево-судинної системи. Це єдиний спосіб гарантувати успішну вагітність та безпечне народження дитини.
Щоб уникнути негативних наслідків, пацієнти з арахнодактилією повинні регулярно проходити обстеження у лікарів, запобігати розвитку серцево-судинних та опорно-рухових патологій. Їм рекомендується вести виключно здоровий спосіб життя, з помірною фізичною активністю.
Діагностика арахнодактилії
Клінічна діагностика арахнодактилії включає збір скарг, сімейний анамнез, оцінку фенотипу, а також антропометричні та фізикальні обстеження. Важливо визначити наявність проявів патологій сполучної тканини у прямих родичів: для цього проводиться генеалогічне дослідження. [ 8 ]
Лабораторна діагностика арахнодактилії передбачає вивчення стану певних видів сполучної тканини, представленої кістковою та хрящовою тканиною, кров’ю та лімфою. Найбільш інформативним вважається дослідження рівнів оксипроліну та глікозаміногліканів у добовій кількості сечі. Крім того, оцінюється вміст лізину, проліну та оксипроліну в крові. Для визначення змінених співвідношень різних видів колагену та структурних аномалій колагенових волокон призначається типування методом непрямої імунофлуоресценції з використанням поліклональних антитіл до колагену та фібронектину. [ 9 ] Залежно від показань, для відображення якості метаболічних процесів сполучної тканини проводяться такі тести:
- глікозаміноглікани, фібрилін, фібронектин;
- вивчення гідроксипроліну, маркерів біосинтезу колагену 1 типу, амінотермінальних пропептидів проколагену 1 типу, маркерів деградації колагену 1 типу, галактозилоксілізину, дезоксипіридиноліну;
- аналіз регуляції метаболізму колагену (вивчення матриксних металопротеїназ та тканинних інгібіторів матриксних металопротеїназ, трансформуючого фактора росту);
- аналіз мікро- та макроелементів (вміст кальцію та магнію, фосфору, заліза, сірки та міді, кобальту та селену, цинку та марганцю, фтору та ванадію, кремнію та бору);
- вивчення маркерів формування кісткової тканини та швидкості процесів ремоделювання (аналіз вмісту остеокальцину, кісткової лужної фосфатази, паратиреоїдного гормону, соматотропного гормону, пролактину, вітаміну D3 , пентозидину, гомоцистеїну в сечі та крові).
Інструментальна діагностика спрямована на виявлення наявних вад розвитку та оцінку функціонального стану органів і систем. Серцево-судинні порушення при арахнодактилії вимагають обов'язкового включення до діагностичного переліку таких процедур, як:
- електрокардіографія;
- Цілодобовий моніторинг ЕКГ;
- ультразвукове дослідження серця та судин.
Судинні аномалії досліджуються за допомогою комп'ютерної томографії або магнітно-резонансної томографії. Додаткові діагностичні процедури можуть включати:
- УЗД органів черевної порожнини, сечостатевої системи;
- Рентген органів грудної клітки, кульшових суглобів; [ 10 ]
- КТ або МРТ хребта.
Після всіх необхідних аналізів пацієнта направляють на генетичну консультацію.
Диференціальна діагностика
Існує фенотипова схожість між синдромом Білса та арахнодактилією при синдромі Марфана (мутації генів FBN2 та FBN1 відповідно), що зумовлено майже повною ідентичністю білкових речовин фібриліну 1 та фібриліну 2. Не дивно, що друга назва синдрому Білса - контрактура арахнодактилії. [ 11 ]
Диференціацію також проводять з іншими патологіями сполучної тканини:
- Синдром Стіклера;
- Синдром Елерса-Данлоса;
- гомоцистинурія;
- артрогрипоз.
Синдром Білса характеризується марфаноїдним фенотипом, вродженими згинальними контрактурами великих і дрібних суглобів, арахнодактилиєю зап'ястя та стопи, викривленням хребта та аномальною формою вуха (так званими «зморщеними» вушними раковинами). Для підтвердження діагнозу проводиться молекулярний аналіз гена FBN2.
Артрогрипоз характеризується множинними контрактурами суглобів, низьким зростом та зниженим інтелектом. Форма пальців та вух нічим не примітна. [ 12 ]
Синдром Елерса-Данлоса характеризується кіфосколіозом, лійкоподібним викривленням грудної клітки, вираженим плоскостопістю та порушенням рефракції. Типовими є підвищена рухливість та вивихи суглобів, висока розтяжність та чутливість шкіри.
Синдром Стіклера характеризується змінами об'єму та скутості суглобів.
До кого звернутись?
Лікування арахнодактилії
Арахнодактилія – це генетична патологія, і наразі медицина не має методів корекції генних дефектів. Тому лікування спрямоване на оптимізацію стану пацієнта, запобігання загостренню патології та усунення симптоматичних проявів. Призначається комплексна терапія, що включає одразу кількох медичних спеціалістів, залежно від типу найбільш виражених симптомів: часто необхідно додатково звертатися до ортопеда, кардіолога, офтальмолога, гастроентеролога та лікарів інших спеціальностей. [ 13 ]
Серед рекомендацій щодо клінічного ведення пацієнтів загальними принципами вважаються наступні:
- обмеження фізичної активності, але не повне її виключення (як частина підтримки серцево-судинної системи);
- медикаментозна терапія;
- за необхідності – хірургічна корекція найбільш уражених ділянок серця та судин;
- ортопедична корекція;
- санаторно-курортне лікування, фізіотерапія, лікувальна фізкультура.
Раціон пацієнтів з арахнодактилією повинен включати достатню кількість високобілкових продуктів, збагачених мікроелементами, вітамінами та жирними кислотами. Рекомендується вживати м'ясо, рибу, морепродукти, бобові та горіхи. Якщо арахнодактилія викликана гомоцистинурією, то споживання тваринного білка різко обмежується.
Дітям худорлявої статури та високого зросту рекомендується з раннього віку вводити в раціон бавовняну та соєву олію, насіння соняшнику, свинячий жир та смалець. Додатково пропонують препарати, що містять поліненасичені жирні кислоти, такі як Омега, які пригнічують вироблення соматотропного гормону.
Для нормалізації білкового обміну призначають вітаміни групи В. Їх також можна отримати з харчових продуктів: гречки, гороху, печінки.
Дуже важливо, щоб аскорбінова кислота надходила в організм пацієнта з їжею. Для цього в раціон обов'язково включають настій шипшини, болгарський перець, капусту, цитрусові, а також обліпиху та цибулю-порей.
За потреби пацієнтам пропонується ортопедична корекція, яка необхідна для зменшення навантаження на хребет і суглоби. Для цього використовується ортопедичне взуття, наколінники та інші апарати, устілки, еластичні бинти.
Хірургічне лікування проводиться за показаннями.
Препарати
Медикаментозне лікування арахнодактилії проводиться 1-2 рази на рік, залежно від стану пацієнта та тяжкості патологічних симптомів. Тривалість лікувального курсу визначається індивідуально та становить у середньому 4 місяці. [ 14 ]
Для стимуляції утворення колагену призначають такі препарати: Піаскледін 300, L-лізин або L-пролін у поєднанні з комплексними полівітамінними препаратами, що містять аскорбінову кислоту, вітаміни групи В, токоферол, магній, цинк, селен, мідь.
Серед хондропротекторів найоптимальнішим є використання хондроїтин сульфату та глюкозаміну сульфату – препаратів, що беруть участь у регуляції метаболізму хондроцитів, пригніченні синтезу ферментів, підвищенні чутливості хондроцитів до ферментативної дії, стимуляції анабілістичних процесів тощо. Оптимальними вважаються комбіновані засоби, такі як Терафлекс, Артрофлекс, Артра тощо.
Мінеральний обмін стимулюють препарати, що нормалізують фосфорно-кальцієві процеси. Препаратами вибору часто є активні форми вітаміну D: Альфа D 3 - Тева, Оксидевіт, Бонвіва тощо. Одночасно використовуються препарати фосфору, кальцію та магнію. Під час лікування приблизно раз на 20 днів перевіряють рівень кальцію та фосфору в крові або сечі, а також проводять аналіз крові на лужну фосфатазу.
Для покращення біоенергетичного стану організму можливе призначення Фосфадену, Рибоксину, Лецитину, Елькар, Коензиму Q10.
Приблизна схема лікування може виглядати так:
- Хондропротектор у віковій дозі, приймати під час їжі та достатньої кількості води. Тривалість одного курсу лікування становить 3-4 місяці.
- L-пролін у дозуванні 500 мг (дітям від 12 років та дорослим) приймають за півгодини до їди, 1-2 рази на день, протягом шести тижнів. За показаннями додатково може бути призначений амінокислотний комплекс – L-пролін, L-лізин, L-лейцин у кількості 10-12 мг/кілограм ваги. Прийом здійснюється 1-2 рази на день, протягом двох місяців.
- Вітамінно-мінеральні комплексні препарати Центрум, або Вітрум, або Юнікап, у дозуванні з урахуванням віку. Тривалість прийому – 4 тижні.
Така схема лікування доцільна, якщо пацієнт з арахнодактилиєю скаржиться на проблеми з опорно-руховим апаратом, а результати аналізів свідчать про підвищене виділення глікозаміногліканів у добовій сечі та зниження рівня вільних амінокислот у крові.
Як правило, лікування добре переноситься пацієнтами, без особливих побічних ефектів. Якщо виявляються реакції гіперчутливості, препарати замінюють, а схему лікування коригують.
Фізіотерапевтичне лікування
Фізіотерапевтичні процедури при арахнодактилії призначаються за показаннями. Наприклад, при недостатньому остеогенезі, для покращення загоєння переломів кісток або при ознаках остеопорозу рекомендується електрофорез з 5% розчином хлориду кальцію, 4% розчином сульфату магнію, 2% розчином сульфату міді або 2% розчином сульфату цинку.
Якщо виявлено вегето-судинні порушення, використовується 1% розчин кофеїну бензоату натрію, ефедрину гідрохлорид або мезатон.
Для активації функціонування кори надниркових залоз призначають медикаментозний електрофорез з 1,5% етимізолом та дециметрову терапію на область надниркових залоз. [ 15 ]
Для стабілізації тонусу судин рекомендуються водні процедури, що сприяють судинній «гімнастиці». Чудово підходять ванни, такі як вуглекисла, хвойна, хлоридно-воднева, сірководнева, радонова. Практикуються обтирання, обливання, контрастний душ, пінні та сольові ванни.
Для покращення стану хрящової тканини використовується магнітотерапія, індуктотерапія, лазеротерапія, електрофорез з диметилсульфоксидом (Димексид).
Хірургічне лікування
Хірургічні операції при арахнодактилії призначаються суворо за показаннями. Наприклад, якщо виявляються тяжкі гемодинамічні порушення на тлі пролапсу стулок клапанів, проводиться масивна аневризма аорти, заміна клапана та пошкодженого сегмента аорти. [ 16 ]
Якщо є виражені функціональні порушення дихальної та серцево-судинної систем, що виникають внаслідок сильного викривлення грудної клітки, проводиться торакопластика.
У разі прогресуючого больового синдрому, спричиненого тяжким сколіозом 3-4 ступеня, також показано хірургічне втручання. З офтальмологічної точки зору абсолютними показаннями до видалення кришталика є субвивих кришталика, ускладнений вторинною глаукомою, а також катаракта та дегенеративні зміни сітківки з високим ризиком відшарування.
Будь-яка операція пацієнтам з арахнодактилією та іншими порушеннями, що вражають структури сполучної тканини, проводиться лише на етапі відносного клінічного та біохімічного полегшення. Після втручання обов'язкове тривале медичне спостереження та інтенсивна терапія препаратами, що покращують метаболізм сполучної тканини.
Профілактика
Арахнодактилія – рідкісна генетична патологія. Іноді вона виникає спонтанно у, здавалося б, здорових батьків. Тому запобігти захворюванню заздалегідь дуже і дуже важко.
Якщо у одного з потенційних батьків обтяжений сімейний анамнез – тобто відомо, що хтось у родині страждав на арахнодактилію – то подружжю слід відвідати генетика та пройти генетичне обстеження на етапі планування дитини. Після підтвердження вагітності лікар проводить пренатальну діагностику, що включає ультразвукове дослідження плода та ряд біохімічних аналізів:
- аналіз крові матері;
- аналіз амніотичної рідини;
- біопсія ворсин хоріона;
- аналіз клітин плаценти та пуповинної крові.
Пацієнти з арахнодактилією перебувають під медичним наглядом протягом усього життя. Профілактичні заходи у пацієнтів спрямовані на запобігання ускладненням. З цією метою проводиться кардіохірургічна корекція, призначається медикаментозне лікування для усунення ризику утворення тромбів. Періодично проводяться курси антибіотикотерапії.
Прогноз
Прогноз для життя пацієнтів з арахнодактилією насамперед визначається тяжкістю супутніх розладів – зокрема, захворювань серцево-судинної, опорно-рухового апарату та органів зору. Найчастіше патологія ускладнюється розривом та розшаруванням аорти. Це необхідно враховувати та своєчасно проводити кардіохірургічну корекцію, що допоможе продовжити життя пацієнта та покращити його якість.
Якщо діагноз був поставлений своєчасно, то за умови дотримання рекомендацій лікаря прогноз можна назвати умовно сприятливим. Медична підтримка значно полегшує перебіг арахнодактилії, і пацієнти мають можливість жити нормальним і відносно повноцінним життям без розвитку серйозних ускладнень.