Медичний експерт статті

Нові публікації
Мукополісахаридоз 3 типу
Останній перегляд: 04.07.2025

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
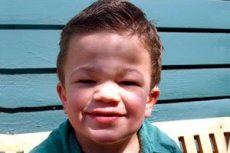
Мукополісахаридоз, тип III (синоніми: синдром Санфіліппо, лізосомний дефіцит α-ацетилглюкозамінідази - мукополісахаридоз III A, дефіцит ацетил-КоА-α-глюкозамінід-N-ацетилтрансферази - мукополісахаридоз III B, N-ацетилглюкозамін-6-сульфатаза - мукополісахаридоз III C, дефіцит сульфамідази - мукополісахаридоз III D).
 [
[ Патогенез
Захворювання спричинене мутаціями в чотирьох різних генах: лізосомальній α-ацетилглюкозамінідазі (мукополісахаридоз III A), ацетил-КоА-α-глюкозамінід-N-ацетилтрансферазі (мукополісахаридоз III B), лізосомальній N-ацетилглюкозамін-6-сульфатазі (мукополісахаридоз III C) та сульфамідазі (мукополісахаридоз III D). Усі ферменти беруть участь у метаболізмі гепарансульфату.
Ген гепаран-N-сульфатази – SGSH – розташований на довгому плечі 17-ї хромосоми – 17q25.3. 75,3% відомих на даний момент мутацій у гені SGSH є точковими. Описано часто зустрічаються мутації, характерні для європейських популяцій – R74C (56% у Польщі та 21% у Німеччині) та R245H (56% у Нідерландах).
Частота мутації R74C становить 47,5%, мутації R245H – 7,5%. Дві інші описані мутації, delll35G та N389S, разом становлять 21,7% мутантних алелів.
Ген aN-ацетилглюкозамінідази (NAGLU) розташований на довгому плечі 17-ї хромосоми - 17q21. 69% мутацій, виявлених у гені NAGLU, є місенс- та нонсенс-мутаціями, 26,3% - невеликими делеціями та інсерціями. Ген ацетил-CoA-cc-глюкозамінід-N-ацетилтрансферази (HGSNAT) розташований на короткому плечі 8-ї хромосоми - 8p11.1. Ген був охарактеризований лише у 2006 році, і на сьогоднішній день у ньому виявлено лише кілька мутацій.
Ген N-ацетилглюкозамін-6-сульфатази – GNS – розташований на довгому плечі 12-ї хромосоми – 12ql4. У світі зареєстровано 12 пацієнтів з мукополісахаридозом IIID. Описано чотири мутації в гені GNS.
При всіх підтипах мукополісахаридозу III спостерігається порушення деградації гепарансульфату, який входить до складу клітинних мембран, включаючи нейрональні, що корелює з важким нейродегенеративним процесом, спричиненим кортикальною атрофією. Хронічна діарея пояснюється залученням вегетативної нервової системи до патологічного процесу разом із порушенням функції слизової оболонки кишечника. Сенсоневральна приглухуватість, ймовірно, зумовлена трьома причинами: частими отитами, деформацією слухових кісточок та аномаліями внутрішнього вуха. Скутість суглобів є результатом деформації метафізів, потовщення суглобової капсули є вторинним по відкладенню глікозаміногліканів та фіброзу. Внутрішньосиндромальні відмінності у тяжкості захворювання зумовлені виключно залишковою функціональною активністю мутантного ферменту: чим вона вища, тим легше протікає захворювання.
Симптоми мукополісахаридозу 3 типу
Клінічний поліморфізм при синдромі Санфіліппо менш виражений, ніж при інших типах мукополісахаридозу. Характерні повільне прогресування захворювання, тяжкі неврологічні порушення з незначними симптомами з боку внутрішніх органів та інших систем.
Перші симптоми захворювання зазвичай з'являються у віці від 2 до 6 років у дітей з раніше нормальним розвитком. До маніфестних симптомів належать регрес психомоторного та мовленнєвого розвитку, психічні розлади у вигляді синдрому гіперактивності, аутична або агресивна поведінка, порушення сну; діти стають необережними та неуважними.
Іншими поширеними симптомами є гірсутизм, жорстке волосся, помірна гепатоспленомегалія, вальгусна деформація кінцівок та коротка шия. Розвиток грубих рис обличчя, таких як гаргоїлізм, та деформацій скелета, таких як множинний дизостоз, слабо виражені при мукополісахаридозі III порівняно з іншими типами мукополісахаридозу, що характеризуються фенотипом Гурлера. Зріст, як правило, відповідає віку, а скутість суглобів рідко викликає порушення функції. У більшості пацієнтів часто розвивається остеопороз та остеомаляція. Вторинні порушення скелета – високий ризик патологічних переломів. Тяжкі психоневрологічні розлади найчастіше спостерігаються до 6-10-го року життя, вони призводять до вираженої соціальної дезадаптації. Прогресуюча сенсоневральна приглухуватість властива всім пацієнтам з тяжкими та середньої тяжкості формами захворювання. Судоми спостерігаються майже у всіх пацієнтів у міру прогресування захворювання.
Хвороба швидко прогресує, і більшість пацієнтів не доживають до 20 років. Мукополісахаридоз IIIA вважається найпоширенішим і найважчим типом цього синдрому.
Діагностика мукополісахаридозу 3 типу
Діагноз мукополісахаридозу III підтверджується визначенням рівня екскреції глікозаміногліканів із сечею та вимірюванням активності ферментів. У разі мукополісахаридозу III збільшується загальна екскреція глікозаміногліканів із сечею та спостерігається гіперекскреція гепарансульфату. Активність лізосомальних ферментів, що відповідають певному підтипу мукополісахаридозу III, вимірюється в лейкоцитах або культурі фібробластів шкіри за допомогою штучного флуорогенного субстрату.
Пренатальна діагностика можлива шляхом вимірювання активності ферментів у біопсії ворсин хоріона на 9-11 тижні вагітності та/або визначення спектру глікозаміногліканів в амніотичній рідині на 20-22 тижні вагітності. Для сімей з відомим генотипом ДНК-діагностика може бути проведена на ранніх термінах вагітності.
Які аналізи необхідні?
Диференціальна діагностика
Диференціальну діагностику проводять як у межах групи мукополісахаридозів, так і з іншими хворобами накопичення лізосом: муколіпідозами, галактосіалідозом, сіалідозом, манозидозом, фукозидозом, гангліозидозом GM1.
До кого звернутись?
Лікування мукополісахаридозу 3 типу
На сьогоднішній день ефективних методів терапії мукополісахаридозу III не розроблено. Показана симптоматична терапія.
Использованная литература