Медичний експерт статті

Нові публікації
Т-клітинні лімфоми шкіри
Останній перегляд: 04.07.2025

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
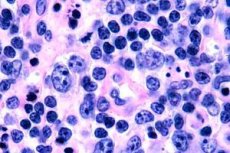
Найчастіше Т-клітинні лімфоми реєструються у людей похилого віку, хоча поодинокі випадки захворювання відзначаються навіть у дітей. Чоловіки хворіють вдвічі частіше, ніж жінки. Т-клітинні лімфоми мають епідермотропний характер.
Причини Т-клітинних лімфом шкіри
Причини та патогенез шкірних Т-клітинних лімфом до кінця не вивчені. Наразі більшість дослідників вважають вірус Т-клітинного лейкозу людини 1 типу (HTLV-1) I основним етіологічним фактором, що ініціює розвиток злоякісних Т-клітинних лімфом шкіри. Поряд з цим обговорюється роль інших вірусів у розвитку Т-клітинної лімфоми: вірусу Епштейна-Барр, простого герпесу 6 типу. У пацієнтів з Т-клітинною лімфомою віруси виявляються в шкірі, периферичній крові та клітинах Лангерганса. Антитіла до HTLV-I виявляються у багатьох пацієнтів з грибоподібним мікозом.
Важливе місце в патогенезі Т-клітинних лімфом відіграють імунопатологічні процеси в шкірі, основним з яких є неконтрольована проліферація клональних лімфоцитів.
Цитокіни, що продукуються лімфоцитами, епітеліальними клітинами та клітинами макрофагальної системи, мають прозапальну та проліферативну дію (IL-1, що відповідає за диференціацію лімфоцитів; IL-2 - фактор росту Т-клітин; IL-4 та IL-5, що збільшують приплив еозинофілів в осередок ураження та їх активацію тощо). В результаті припливу Т-лімфоцитів в осередок ураження утворюються мікроабсцеси Потьє. Одночасно зі збільшенням проліферації лімфоцитів пригнічується активність клітин протипухлинного захисту: природних кілерів, лімфоцитотоксичних лімфоцитів, дендритних клітин, зокрема, клітин Лангерганса, а також цитокінів (IL-7, IL-15 тощо) - інгібіторів росту пухлини. Не можна виключати роль спадкових факторів. Наявність сімейних випадків, часте виявлення деяких антигенів гістосумісності (HLA B-5 та HLA B-35 – при високозлоякісних лімфомах шкіри, HLA A-10 – при менш агресивних лімфомах, HLA B-8 – при еритродермічній формі грибоподібного мікозу) підтверджують спадкову природу дерматозу.
Клінічні спостереження вказують на можливу трансформацію тривало перебігаючих хронічних дерматозів (нейродерміт, атопічний дерматит, псоріаз тощо) у грибоподібний мікоз. Ключовим фактором є тривале персистентування лімфоцитів у вогнищі запалення, які порушують імунний нагляд та сприяють появі клону злоякісних лімфоцитів і, таким чином, розвитку злоякісного проліферативного процесу.
Вплив фізичних факторів на організм, таких як інсоляція, іонізуюче випромінювання, хімічні речовини, може призвести до появи клону «генотравматичних» лімфоцитів, що мають мутагенну дію на лімфоїдні клітини, та розвитку злоякісного новоутворення лімфоцитів.
Отже, Т-клітинні лімфоми можна розглядати як багатофакторне захворювання, яке починається з активації лімфоцитів під впливом різних канцерогенних, «генотравматизуючих» факторів та виникнення домінантного клону Т-клітин. Тяжкість порушення імунного нагляду, клон злоякісних лімфоцитів визначає клінічні прояви (плямистих, бляшкоподібних або пухлинних елементів) Т-клітинних лімфом.
Патогенез
На ранній стадії грибоподібного мікозу спостерігаються акантоз з широкими відростками, гіперплазія та ущільнення базальних кератиноцитів, вакуолярна дегенерація деяких базальних клітин, атипові мітози в різних шарах епідермісу, епідермотропізм інфільтрату з проникненням лімфоцитів в епідерміс. У дермі навколо судин спостерігаються дрібні інфільтрати, що складаються з поодиноких мононуклеарних клітин з гіперхромними ядрами - "мікотичні" клітини. На другій стадії спостерігається наростання вираженості дермального інфільтрату та епідермотропізму клітин інфільтрату, внаслідок чого злоякісні лімфоцити проникають в епідерміс, утворюючи скупчення у вигляді мікроабсцесів Потріє. На третій, пухлинній стадії, спостерігається масивний акантоз та незначна атрофія епідермісу, а також посилена інфільтрація епідермісу пухлинними лімфоцитами, які утворюють множинні мікроабсцеси Потріє. Масивний інфільтрат розташований по всій товщі дерми та покриває частину гіподерми. Спостерігаються бластні форми лімфоцитів.
Шкірна великоанапластична Т-клітинна лімфома
Він представлений групою лімфопроліферативних процесів, що характеризуються наявністю проліфератів з атипових клональних великих анапластичних CD30+ Т-клітин. Як правило, розвивається вторинно на стадії пухлини грибоподібного мікозу або при синдромі Сезарі, але може розвиватися самостійно або з дисемінацією системних лімфом цього типу. Клінічно такі лімфоми відповідають так званій декапітованій формі грибоподібного мікозу у вигляді одиничних або множинних вузлів, зазвичай згрупованих.
Гістологічно проліферат займає майже всю дерму з епідермотропізмом або без нього у випадку епідермальної атрофії.
Цитологічно пухлинні клітини можуть відрізнятися за розміром і формою. На основі цих властивостей розрізняють середньо- та великоклітинну плеоморфну Т-клітинну лімфому з ядрами різної неправильної конфігурації – звивистими, багатолопатевими, з щільним хроматином, чітко окресленим ядерцем та досить рясною цитоплазмою; імунобластичні – з великими круглими або овальними ядрами з прозорою каріоплазмою та одним центрально розташованим ядерцем; анапластичні – з потворними дуже великими клітинами з ядрами неправильної конфігурації та рясною цитоплазмою. Фенотипово вся ця група належить до Т-хелперних лімфом і може бути CD30+ або CD30-.
Р. Віллемзе та ін. (1994) показали, що перебіг CD30+ лімфоми є більш сприятливим. Генотипово виявляється клональна перебудова рецептора Т-лімфоцитів.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
Симптоми Т-клітинних лімфом шкіри
Найпоширенішим захворюванням у групі Т-клітинних лімфом шкіри є грибоподібний мікоз, на який припадає близько 70% випадків. Розрізняють три клінічні форми захворювання: класичну, еритродермічну та обезголовлену. Т-клітинні лімфоми характеризуються поліморфізмом висипань у вигляді плям, бляшок, пухлин.
Еритродермічна форма грибоподібного мікозу зазвичай починається з неконтрольованого свербіння, набряку, універсальної гіперемії, появи еритематозно-сквамозних уражень на шкірі тулуба та кінцівок, які мають тенденцію до злиття та розвитку еритродермії протягом 1-2 місяців. Майже у всіх пацієнтів спостерігається долонно-підошовний гіперкератоз та дифузне витончення волосся по всій шкірі. Всі групи лімфатичних вузлів значно збільшені. Збільшені пахові, стегнові, пахвові, ліктьові лімфатичні вузли пальпуються у вигляді «пакетів» щільно-еластичної консистенції, не зрощені з навколишніми тканинами, безболісні. Загальний стан різко погіршується: виникає лихоманка з температурою тіла до 38-39°C, нічна пітливість, слабкість та втрата ваги. Наразі синдром Сезарі багатьма дерматологами вважається найрідкіснішим лейкемічним варіантом еритродермічної форми грибоподібного мікозу,
На лімфоцитограмах відзначається виражений лейкоцитоз – клітини Сезарі. Клітини Сезарі – це злоякісні Т-хелпери, ядра яких мають складчасту мозочкову поверхню з глибокими інвагінаціями ядерної оболонки. Летальний результат відзначається через 2-5 років, частою причиною якого є серцево-судинна патологія та інтоксикація.
Декапітована форма грибоподібного мікозу характеризується швидким розвитком пухлиноподібних уражень на, здавалося б, здоровій шкірі без попереднього тривалого утворення бляшок. Ця форма характеризується високим ступенем злоякісності, що вважається проявом лімфосаркоми. Летальний результат спостерігається протягом року.
Стадії
Класична форма грибоподібного мікозу характеризується трьома стадіями розвитку: еритематозно-плоскоклітинною, бляшковою та пухлинною.
Перша стадія нагадує клінічну картину деяких доброякісних запальних дерматозів – екземи, себорейного дерматиту, бляшкового парапсоріазу. На цій стадії захворювання спостерігаються плями різного розміру, інтенсивно рожевого, рожево-червоного з фіолетовим відтінком кольору, круглих або овальних обрисів, з відносно чіткими межами, поверхневим висівкоподібним або дрібнопластиновим лущенням. Елементи часто розташовуються на різних ділянках шкіри, найчастіше на тулубі та обличчі. Поступово їх кількість збільшується. З часом процес може набути характеру еритродермії (еритродермічна стадія). Висип може існувати роками або спонтанно зникати. На відміну від доброякісних запальних дерматозів, елементи висипу та свербіж на цій стадії стійкі до терапії.
Інфільтративно-бляшкова стадія розвивається протягом кількох років. На місці раніше існуючих плямистих висипань з'являються бляшки круглих або неправильних контурів, інтенсивно-фіолетового кольору, чітко відмежовані від здорової шкіри, щільні, з лущиться поверхнею. Їх консистенція нагадує «товстий картон». Деякі з них спонтанно розсмоктуються, залишаючи ділянки темно-коричневої гіперпігментації та/або атрофії (пойкілодермія). Свербіж на цій стадії ще інтенсивніший та болючіший, спостерігається лихоманка та втрата ваги. На цій стадії може спостерігатися лімфаденопатія.
На третій, пухлинній стадії, з'являються безболісні пухлини щільної, еластичної консистенції, жовто-червоного кольору, що розвиваються з бляшок або виникають на, здавалося б, здоровій шкірі. Форма пухлин куляста або сплющена, часто нагадує капелюшок гриба. Пухлини можуть з'являтися будь-де. Їх кількість коливається в широких межах від одиниць до десятків, розміри - від 1 до 20 см у діаметрі. При розпаді тривало існуючих пухлин утворюються виразки з нерівними краями та глибоким дном, що досягають фасції або кістки. Найчастіше уражаються лімфатичні вузли, селезінка, печінка та легені. Загальний стан погіршується, з'являються та наростають симптоми інтоксикації, розвивається слабкість. Середня тривалість життя пацієнтів з класичною формою грибоподібного мікозу з моменту постановки діагнозу становить від 5 до 10 років. Смертність зазвичай спостерігається від інтеркурентних захворювань: пневмонії, серцево-судинної недостатності, амілоїдозу. Суб'єктивно відчувається свербіж, а при розпаді пухлин - біль в уражених ділянках.
Що потрібно обстежити?
Як обстежувати?
Лікування Т-клітинних лімфом шкіри
На еритематозно-сквамозній стадії пацієнтам не потрібна протипухлинна терапія; їм призначають місцеві кортикостероїди (преднізолон, бетаметазон, похідні дексаметазону), інтерферон альфа (3 млн МО щодня, потім 3 рази на тиждень протягом 3-6 місяців залежно від клінічних проявів або ефективності лікування), інтерферон гамма (100 000 МО на день протягом 10 днів, цикл повторюють 12-3 рази з 10-денною перервою), ПУВА-терапію або Re-ПУВА-терапію. Ефективність ПУВА-терапії базується на селективному утворенні ковалентних зшивок псораленів з ДНК у проліферуючих Т-хелперах, що пригнічує їх поділ. На другій стадії, крім вищезазначених засобів, використовуються системні кортикостероїди (30-40 мг преднізолону на день протягом 1,5-2 місяців) та цитостатики (проспедин 100 мг на день щодня, всього 4-5 ін'єкцій). Поєднання інтерферонів з іншими методами терапії має більш виражений терапевтичний ефект (інтерферони + ПУВА, інтерферони + цитостатики, інтерферони + ароматичні ретиноїди).
У стадії пухлини основним методом є поліхіміотерапія. Використовується комбінація вінкристину (0,5-1 мг внутрішньовенно один раз на день, всього 4-5 ін'єкцій) з преднізолоном (40-60 мг на день перорально під час хіміотерапії), проспідином (100 мг на день, всього 3 г) та інтерферонами. Рекомендується фотодинамічна, електронно-променева терапія та фотоферез (екстракорпоральна фотохіміотерапія).