Медичний експерт статті

Нові публікації
Вірус імунодефіциту людини (ВІЛ)
Останній перегляд: 04.07.2025

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
Синдром набутого імунодефіциту був визначений як специфічне захворювання в 1981 році в Сполучених Штатах, коли у низки молодих людей виникли серйозні захворювання, спричинені мікроорганізмами, непатогенними або слабопатогенними для здорових людей. Дослідження імунного статусу пацієнтів виявило різке зниження кількості лімфоцитів загалом і Т-хелперів зокрема. Цей стан отримав назву СНІД (синдром набутого імунодефіциту). Спосіб зараження (статевий контакт, через кров та її препарати) вказував на інфекційну природу захворювання.
Збудника СНІДу незалежно відкрили у 1983 році француз Л. Монтаньє, який назвав його вірусом, асоційованим з лімфоаденопатією LAV, оскільки виявив його у пацієнта з лімфаденопатією; та американець Р. Галло, який назвав вірус HTLV-III (Human T-lymphotropic Virus III): раніше він відкрив лімфотропні віруси I та II.
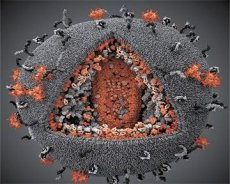
Порівняння властивостей вірусів LAV та HTLV-III показало їхню ідентичність, тому, щоб уникнути плутанини, у 1986 році вірус був названий ВІЛ (вірус імунодефіциту людини або ВІЛ). ВІЛ має сферичну форму, діаметром 110 нм. Оболонка вірусу має форму багатогранника, що складається з 12 п'ятикутників та 20 шестикутників. У центрі та кутах кожного шестикутника знаходиться молекула глікозильованого білка gpl20 (число 120 вказує на молекулярну масу білка в кілодальтонах). Загалом на поверхні віріона розташовано 72 молекули gpl20 у вигляді своєрідних шипів, кожен з яких пов'язаний з внутрішньомембранним білком gp41. Ці білки разом з подвійним ліпідним шаром утворюють суперкапсид (мембрану) віріона.
Білки gpl20 та gp41 утворюються шляхом розрізання білка-попередника Env клітинною протеазою. Білок gp41 формує шипоподібний «стебло», зв'язуючись своїм цитоплазматичним доменом з матриксним білком p17MA, розташованим безпосередньо під оболонкою. Молекули p17 взаємодіють під час дозрівання віріона, утворюючи ікосаедр, що лежить під оболонкою.
У центральній частині віріона білок p24 утворює конусоподібний капсид. Звужена частина капсиду з'єднана з мембраною віріона за участю білка rb. Усередині капсиду знаходяться дві однакові молекули вірусної геномної РНК. Вони з'єднані своїми 5'-кінцями з нуклеокапсидним білком p7NC. Цей білок цікавий тим, що має два амінокислотні залишки (мотиви), багаті на цистеїн та гістидин і містять атом Zn - їх називають "цинковими пальцями", оскільки вони захоплюють молекули геномної РНК для включення до складу віріонів, що формуються. Капсид також містить три ферменти. Ревертаза (RT), або pol-комплекс, включає зворотну транскриптазу, РНКазу H та ДНК-залежну ДНК-полімеразу. Ревертаза присутня у вигляді гетеродимера p66/p51. Протеаза (PR) - p10, ініціює та реалізує процес дозрівання віріона. Інтеграза (IN) - p31, або ендонуклеаза, забезпечує включення провірусної ДНК у геном клітини-хазяїна. Капсид також містить молекулу праймерної РНК (тРНК1"3).
РНК-геном у клітині за допомогою зворотної транскриптази перетворюється на ДНК-геном (ДНК-провірус), що складається з 9283 пар нуклеотидів. Він обмежений зліва та справа так званими довгими кінцевими повторами, або LTR: S'-LTR зліва та 3'-LTR справа. LTR містять по 638 пар нуклеотидів кожен.
Геном ВІЛ складається з 9 генів, деякі з яких перекриваються на кінцях (мають кілька рамок зчитування) та мають екзонинтронну структуру. Вони контролюють синтез 9 структурних та 6 регуляторних білків.
Важливість LTR для вірусного геному полягає в тому, що вони містять такі регуляторні елементи, які контролюють його функціонування:
- транскрипційний сигнал (промоторна ділянка);
- сигнал додавання полі-А;
- сигнал обмеження;
- сигнал інтеграції;
- сигнал позитивної регуляції (TAR для білка TAT);
- негативний регуляторний елемент (NRE для білка NEF);
- сайт для приєднання праймера РНК (тРНК™3) для синтезу мінус-ланцюга ДНК на 3'-кінці; сигнал на 5'-кінці LTR, який служить праймером для синтезу плюс-ланцюга ДНК.
Крім того, LTR містить елементи, що беруть участь у регуляції сплайсингу мРНК, упаковці молекул vRNA в капсид (Psi-елемент). Нарешті, під час транскрипції геному в довгих мРНК формуються два сигнали для білка REV, які перемикають синтез білка: CAR – для регуляторних білків та CRS – для структурних білків. Якщо білок REV зв'язується з CAR, синтезуються структурні білки; якщо він відсутній, синтезуються лише регуляторні білки.
Наступні регуляторні гени та їхні білки відіграють особливо важливу роль у регуляції функціонування вірусного геному:
- Білок TAT, який здійснює позитивний контроль реплікації вірусу та діє через регуляторну область TAR;
- Білки NEV та VPU, які здійснюють негативний контроль реплікації через область NRE;
- Білок REV, який здійснює позитивно-негативний контроль. Білок REV контролює роботу генів gag, pol, env та здійснює негативну регуляцію сплайсингу.
Таким чином, реплікація ВІЛ знаходиться під потрійним контролем – позитивним, негативним та позитивно-негативним.
Білок VIF визначає інфекційність новосинтезованого вірусу. Він пов'язаний з капсидним білком p24 та присутній у віріоні в кількості 60 молекул. Білок NEF представлений у віріоні невеликою кількістю молекул (5-10), можливо, пов'язаних з оболонкою.
Білок VPR пригнічує клітинний цикл у фазі G2, бере участь у транспортуванні преінтеграційних комплексів у ядро клітини, активує деякі вірусні та клітинні гени, а також підвищує ефективність реплікації вірусу в моноцитах і макрофагах. Розташування білків VPR, TAT, REV та VPU у віріоні не встановлено.
Окрім власних білків, мембрана віріона може містити деякі білки клітини-хазяїна. Білки VPU та VPR беруть участь у регуляції розмноження вірусу.
Антигенні варіанти вірусу імунодефіциту людини (ВІЛ)
Вірус імунодефіциту людини (ВІЛ) дуже мінливий. Навіть з організму одного пацієнта можна виділити штами вірусу, які суттєво відрізняються за антигенними властивостями. Така мінливість сприяє інтенсивному руйнуванню CD4+ клітин та потужній антитільній відповіді на ВІЛ-інфекцію. У пацієнтів із Західної Африки виділено нову форму ВІЛ – ВІЛ-2, біологічно близьку до ВІЛ-1, але імунологічно відмінну від нього. Гомологія первинної структури геномів цих вірусів становить 42%. ДНК-провірус ВІЛ-2 містить 9671 п.н., а його LTR – 854 п.н. Згодом ВІЛ-2 був виділений і в інших регіонах світу. Перехресного імунітету між ВІЛ-1 та ВІЛ-2 немає. Відомі дві великі форми ВІЛ-1: О (Випадок) та М (Основна), остання поділяється на 10 підтипів (AJ). У Росії циркулює вісім підтипів (AH).
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
Механізм взаємодії ВІЛ з клітиною
Проникнувши в організм, вірус спочатку атакує клітини, що містять специфічний для нього рецептор CD4. Цей рецептор присутній у великій кількості в Т-хелперах, у меншій кількості - в макрофагах і моноцитах, причому Т-хелпери особливо чутливі до вірусу.
Вірус імунодефіциту людини (ВІЛ) розпізнає рецептори CD4 за допомогою свого білка gpl20. Процес взаємодії ВІЛ з клітиною відбувається за такою схемою: рецептор-опосередкована адсорбція -> покрита ямка -> покрита везикула -> лізосома. У ній мембрана віріона зливається з мембраною лізосоми, і нуклеокапсид, звільнившись від суперкапсиду, потрапляє в цитоплазму; на шляху до ядра він руйнується, а геномна РНК та пов'язані з нею компоненти ядра вивільняються. Потім зворотна транскриптаза синтезує мінус-ланцюг ДНК на РНК віріона, потім РНКаза H руйнує РНК віріона, а вірусна ДНК-полімераза синтезує плюс-ланцюг ДНК. На кінцях ДНК-провірусу утворюються 5'-LTR та 3'-LTR. ДНК-провірус може деякий час залишатися в ядрі в неактивній формі, але рано чи пізно він інтегрується в хромосому клітини-мішені за допомогою своєї інтегрази. У ньому провірус залишається неактивним доти, доки даний Т-лімфоцит не буде активований мікробними антигенами або іншими імунокомпетентними клітинами. Активація клітинної транскрипції ДНК регулюється спеціальним ядерним фактором (NF-kB). Це ДНК-зв'язуючий білок і виробляється у великих кількостях під час активації та проліферації Т-лімфоцитів і моноцитів. Цей білок зв'язується з певними послідовностями клітинної ДНК та подібними LTR-послідовностями ДНК-провірусу та індукує транскрипцію як клітинної ДНК, так і ДНК-провірусу. Індукуючи транскрипцію ДНК-провірусу, він здійснює перехід вірусу з неактивного стану в активний і, відповідно, від персистуючої інфекції до продуктивної. Провірус може залишатися в неактивному стані дуже довго. Активація вірусу є критичним моментом у його взаємодії з клітиною.
З моменту проникнення вірусу в клітину починається період ВІЛ-інфекції – стан вірусоносійства, який може тривати 10 років і більше; а з моменту активації вірусу починається захворювання – СНІД. За допомогою своїх регуляторних генів та їх продуктів вірус починає активно розмножуватися. Білок TAT може збільшити швидкість розмноження вірусу в 1000 разів. Вірусна транскрипція є складною. Вона включає формування як повнорозмірної, так і субгеномної мРНК, сплайсинг мРНК, а потім відбувається синтез структурних та регуляторних білків.
Синтез структурних білків відбувається наступним чином. Спочатку синтезується поліпротеїн-попередник Pr55Gag (білок з молекулярною масою 55 кДа). Він містить 4 основні домени: матриксний (MA), капсидний (CA), нуклеокапсидний (NC) та домен rb, з якого в результаті розрізання Pr55Gag вірусною протеазою (він самовідщеплюється від іншого білка-попередника, Gag-Pol), утворюються структурні білки p17, p24, p7 та rb відповідно. Утворення поліпротеїну Pr55Gag є основною умовою формування вірусних частинок. Саме цей білок визначає програму морфогенезу віріона. Вона послідовно включає етапи транспортування поліпротеїну Gag до плазматичної мембрани, взаємодії з нею та білок-білкових взаємодій під час формування вірусної частинки та її брунькування. Pr55Gag синтезується на вільних полірибосомах; молекули білка транспортуються до мембрани, де вони закріплюються своїми гідрофобними ділянками. CA-домен відіграє основну роль у створенні нативної конформації білка Gag. NC-домен забезпечує включення (за допомогою своїх «цинкових пальців») 2 молекул геномної РНК до складу вірусної частинки, що формується. Молекула поліпротеїну спочатку димеризується завдяки взаємодії матричних доменів. Потім димери об'єднуються в гексамерні (з 6 одиниць) комплекси в результаті взаємодії CA- та NC-доменів. Зрештою, гексамери, з'єднуючись на своїх бічних поверхнях, утворюють незрілі сферичні віріони, всередині яких міститься геномна вірусна РНК, захоплена NC-доменом.
Інший білок-попередник, Prl60Gag-Pol (білок з молекулярною масою 160 кДа), синтезується в результаті зсуву рамки зчитування рибосомою під час трансляції 3'-кінця гена gag в області, розташованій безпосередньо перед областю, що кодує білок rb. Цей поліпротеїн Gag-Pol містить неповну послідовність білка Gag (1-423 амінокислоти) та послідовності Pol, що включають домени PR, RT та IN. Молекули поліпротеїну Gag-Pol також синтезуються на вільних полірибосомах і транспортуються до плазматичної мембрани. Поліпротеїн Prl60Gagpol містить усі сайти міжмолекулярної взаємодії та сайти зв'язування з мембраною, властиві поліпротеїну Gag. Тому молекули поліпротеїну Gag-Pol зливаються з мембраною і разом з молекулами Gag включаються до складу віріонів, що формуються, що призводить до появи активної протеази та початку процесу дозрівання віріона. Протеаза ВІЛ-1 має високу активність лише у формі димеру, тому для її самовиділення з Prl60Gag-Pol необхідна димеризація цих молекул. Дозрівання віріона полягає в тому, що вивільнена активна протеаза розрізає prl60Gag-Pol та Gag55 у розпізнаваних нею сайтах; утворюються білки p17, p24, p7, p6, ревертаза, інтеграза та відбувається їх асоціація у вірусну структуру.
Білок Env синтезується на рибосомах, пов'язаних з мембранами ендоплазматичного ретикулуму, потім він глікозилується, розрізається клітинною протеазою на gp120 та gp41 та транспортується на поверхню клітини. У цьому випадку gp41 проникає через мембрану та зв'язується з матриксними доменами молекули білка Gag, пов'язаної з внутрішньою поверхнею мембрани. Цей зв'язок зберігається і в зрілому віріоні.
Таким чином, збірка вірусних частинок полягає в агрегації білків-попередників та пов'язаних з ними молекул РНК на плазматичній мембрані клітини-хазяїна, утворенні незрілих віріонів та їх вивільненні шляхом брунькування з поверхні клітини. Під час брунькування віріон оточує себе клітинною мембраною, в яку вбудовуються молекули gp41 та gp120. Під час брунькування або, можливо, після вивільнення віріонів відбувається їх дозрівання, яке здійснюється за допомогою вірусної протеази та полягає в протеолітичному розрізанні білків-попередників Pr55Gag та Prl60Gag-Pol на білки зрілого вірусу та їх об'єднанні в певні структурні комплекси. Провідну роль у процесах вірусного морфогенезу відіграє поліпротеїн-попередник Pr55Gag, який організовує та збирає незрілий віріон; процес його дозрівання завершується специфічною вірусною протеазою.
Причини імунодефіциту
Однією з основних причин імунодефіциту при ВІЛ-інфекції є масова загибель Т-хелперів. Вона відбувається в результаті наступних подій. По-перше, Т-хелпери, інфіковані вірусом, гинуть внаслідок апоптозу. Вважається, що у хворих на СНІД реплікація вірусу, апоптоз і зменшення кількості Т-хелперів взаємопов'язані. По-друге, Т-кілери розпізнають і знищують Т-клітини, інфіковані вірусом або несучі адсорбовані молекули gpl20, а також інфіковані та неінфіковані вірусом Т-хелпери, які утворюють симпласти (синцитії), що складаються з кількох десятків клітин (деякі з них гинуть в результаті розмноження в них вірусів). В результаті знищення великої кількості Т-хелперів відбувається зниження експресії мембранних рецепторів у В-лімфоцитах до інтерлейкіну-2, порушується синтез різних інтерлейкінів (факторів росту та диференціації В-лімфоцитів - IL-4, IL-5, IL-6 тощо), що призводить до порушення функції системи Т-кілерів. Пригнічується активність систем комплементу та макрофагів. Макрофаги та моноцити, інфіковані вірусом, довго не гинуть, але вони не здатні виводити вірус з організму. Нарешті, завдяки структурній та антигенній подібності gpl20 з рецепторами деяких епітеліальних клітин організму (включаючи рецептори трофобласта, що опосередковують трансплантаційну передачу ВІЛ), синтезуються антирецепторні антитіла широкого спектру дії. Такі антитіла здатні блокувати різні клітинні рецептори та ускладнювати перебіг захворювання аутоімунними порушеннями. Наслідком ВІЛ-інфекції є ураження всіх основних ланок імунної системи. Такі пацієнти стають беззахисними перед широким спектром мікроорганізмів. Це призводить до розвитку опортуністичних інфекцій та пухлин. Для пацієнтів з ВІЛ-інфекцією підвищується ризик розвитку щонайменше трьох видів раку: саркома Капоші; карцинома (включаючи рак шкіри); В-клітинна лімфома, яка виникає внаслідок злоякісного переродження В-лімфоцитів. Однак ВІЛ є не тільки лімфоцитотропним, але й нейротропним. Він проникає в клітини центральної нервової системи (астроцити) як шляхом рецептор-опосередкованого ендоцитозу, так і шляхом фагоцитозу інфікованих вірусом лімфобластів астроцитами. При взаємодії вірусу з астроцитами також утворюються симпласти, які сприяють поширенню збудника міжклітинними каналами. Вірус може тривалий час зберігатися в макрофагах і моноцитах, тому вони служать резервуаром і розподільниками його в організмі, маючи можливість проникати в усі тканини. Інфіковані макрофаги відіграють основну роль у впровадженні ВІЛ у центральну нервову систему та її пошкодженні. У 10% пацієнтів первинні клінічні синдроми пов'язані з ураженням центральної нервової системи та проявляються як деменція. Таким чином, для людей, інфікованих ВІЛ, характерні 3 групи захворювань - опортуністичні інфекції,пухлинні захворювання та ураження центральної нервової системи.
Епідеміологія ВІЛ-інфекції
Джерелом ВІЛ-інфекції є лише людина – хвора або вірусоносій. Вірус імунодефіциту людини (ВІЛ) міститься в крові, спермі, цервікальній рідині; у матерів-годувальниць – у грудному молоці. Зараження відбувається статевим шляхом, через кров та її препарати, а також від матері до дитини до, під час та після пологів. Випадки зараження вірусом через їжу, напої та укуси комах не відомі.
Наркоманія сприяє поширенню СНІДу. Захворюваність на ВІЛ зростає з кожним роком. За даними ВООЗ, з 1980 по 2000 рік у світі було інфіковано 58 мільйонів людей. Тільки у 2000 році у світі було інфіковано 5,3 мільйона людей, а 3 мільйони людей померли від СНІДу. Станом на 1 січня 2004 року в Росії було зареєстровано 264 тисячі ВІЛ-інфікованих. Половина людей, інфікованих ВІЛ, помирає протягом 11-12 років з моменту зараження. На початок 2004 року з кожних 100 тисяч громадян Росії близько 180 жили з діагнозом "ВІЛ-інфекція". Прогнозується, що за такого рівня захворюваності загальна кількість ВІЛ-інфікованих у Росії до 2012 року становитиме 2,5-3 мільйони людей. Складність боротьби з ВІЛ-інфекцією залежить від низки причин: по-перше, немає ефективних методів її лікування та специфічної профілактики; по-друге, інкубаційний період ВІЛ-інфекції може перевищувати 10 років. Його тривалість залежить від моменту активації Т-лімфоцита та ДНК-провірусу, що міститься в його хромосомі. Досі незрозуміло, чи кожен, хто інфікований вірусом, приречений на СНІД, чи можливе тривале носійство вірусу без захворювання (що видається малоймовірним). Нарешті, існує кілька вірусів імунодефіциту людини (ВІЛ-1, ВІЛ-2), антигенні відмінності між якими перешкоджають формуванню перехресного імунітету. Відкриття вірусу імунодефіциту мавп (ВІЛ) пролило світло на питання походження ВІЛ. ВІЛ подібний до ВІЛ за організацією геному, але суттєво відрізняється нуклеотидною послідовністю. ВІЛ-2 займає проміжне положення між ВІЛ-1 та ВІЛ за своїми серологічними властивостями, а за нуклеотидною послідовністю ближчий до ВІЛ. У зв'язку з цим В. М. Жданов припустив, що віруси ВІЛ-1, ВІЛ-2 та ВІЛ походять від спільного предка. Можливо, за словами Р. Галло, один із ВІЛ якимось чином потрапив в організм людини, де зазнав низки мутацій, що призвели до появи ВІЛ-1, ВІЛ-2 та інших його форм.
Симптоми ВІЛ-інфекції
Вірус імунодефіциту людини має деякі особливості, які значною мірою визначають патогенез захворювання. Вірус має дуже високу швидкість розмноження, що визначається його регуляторними елементами (в активній стадії синтезується до 5000 віріонів за 5 хвилин). Завдяки наявності білка злиття (gp41) вірус індукує утворення великих синцитіальних структур внаслідок злиття інфікованих та неінфікованих Т-хелперів, що призводить до їх масової загибелі. Молекули білка gpl20, що утворюються у великій кількості, вільно циркулюють у крові та зв'язуються з рецепторами неінфікованих Т-хелперів, внаслідок чого вони також розпізнаються та знищуються Т-кілерами. Вірус може поширюватися міжклітинними каналами від клітини до клітини, в цьому випадку він стає погано доступним для антитіл.
Клінічні критерії ВІЛ-інфекції
У дорослих ВІЛ-інфекція діагностується за наявності щонайменше двох серйозних симптомів у поєднанні з щонайменше одним незначним симптомом та за відсутності інших відомих причин імунодефіциту (рак, вроджений імунодефіцит, сильне голодування тощо). До серйозних симптомів належать:
- втрата ваги на 10% і більше;
- тривалий гарячковий стан, періодичний або постійний;
- хронічна діарея.
Незначні симптоми: постійний кашель, генералізований дерматит, рецидивуючий оперізувальний лишай, кандидоз ротової порожнини та глотки, хронічний простий герпес, генералізована лімфаденопатія. Діагноз СНІДу ставиться за наявності лише саркоми Капоші, криптококового менінгіту, пневмоцистної пневмонії. На клінічну картину захворювання впливає супутня опортуністична інфекція.
Методи культивування вірусу імунодефіциту людини (ВІЛ)
ВІЛ-1 та ВІЛ-2 можна культивувати в клітинах лише одного клону лімфоцитів TCV4 – H9, отриманого з лейкемічних лімфоцитів TCV4. Для цих цілей також можна використовувати моношарові культури клітин астроцитів, в яких ВІЛ-1 добре розмножується. Серед тварин до ВІЛ-1 сприйнятливі шимпанзе.
Стійкість вірусу в зовнішньому середовищі низька. Він гине під впливом сонячного світла та УФ-випромінювання, руйнується при 80 °C протягом 30 хвилин, при обробці загальновживаними дезінфекційними засобами - протягом 20-30 хвилин. Для дезінфекції матеріалу, що містить вірус, необхідно використовувати мікобактерицидні дезінфекційні засоби, оскільки вони ефективні проти мікроорганізмів з найвищою стійкістю.
Лабораторна діагностика ВІЛ-інфекції
Основним методом діагностики вірусоносійства та ВІЛ-інфекції є імуноферментний аналіз. Однак, через те, що gpl20 має структурну та антигенну схожість з рецепторами деяких клітин людини, включаючи рецептори, що транспортують імуноглобуліни через епітеліальні клітини слизових оболонок, в організмі можуть з'являтися антитіла, пов'язані з антитілами проти gpl20. У цьому випадку можливі хибнопозитивні результати ІФА. Тому всі позитивно реагуючі сироватки досліджуваних проходять додатковий аналіз методом імуноблоттингу, або вестерн-блоттингу. Цей метод базується на ідентифікації досліджуваних антитіл після їх електрофоретичного розділення та подальшого тестування з використанням мічених антивидових антитіл. Вірусологічний метод використовується рідко через складність культивування вірусу. Клон лімфоцитів H9 використовується для отримання вірусних антигенів – необхідних компонентів діагностичних тест-систем. Метод СЛР дозволяє виявити вірус на ранній стадії віремії.
Лікування ВІЛ-інфекції
Необхідно знайти або синтезувати препарати, які ефективно пригнічують активність зворотної транскриптази (ревертази) або вірусної протеази. Вони б запобігали утворенню ДНК-провірусу та (або) пригнічували внутрішньоклітинне розмноження вірусу. Сучасна стратегія лікування ВІЛ-інфікованих пацієнтів базується на принципі комбінованого застосування препаратів, що пригнічують вірусну протеазу (один з препаратів) та реверсазу (2 різних препарати) - комбінована (потрійна) терапія. У Росії для лікування ВІЛ-інфікованих пацієнтів рекомендується комбіноване застосування 2 вітчизняних препаратів: фосфазиду та криксивану, які специфічно пригнічують репродукцію ВІЛ на ранніх та пізніх стадіях репродукції, особливо при зниженій активності азидотимідину.
Проблема специфічної профілактики полягає в необхідності створення вакцини, яка б забезпечила формування ефективного клітинного імунітету на основі вірусспецифічних цитотоксичних лімфоцитів без будь-якої значної продукції антитіл. Такий імунітет забезпечують Thl-хелпери. Можливо, що антитіла, включаючи віруснейтралізуючі, не тільки неефективні в придушенні ВІЛ-інфекції, але й на високому рівні пригнічують клітинний імунітет. Тому вакцина проти ВІЛ повинна відповідати, перш за все, двом основним вимогам: а) бути абсолютно безпечною та б) стимулювати активність Т-цитотоксичних лімфоцитів. Вивчається ефективність різних варіантів вакцини, отриманих з убитих (інактивованих) вірусів та з окремих антигенів з високими захисними властивостями. Такі антигени можуть бути як виділені з самих віріонів, так і синтезовані хімічно. Запропоновано вакцину, створену на основі методів генної інженерії. Це рекомбінантний вірус віспи, що несе гени ВІЛ, відповідальні за синтез антигенів з сильними імуногенними властивостями. Питання ефективності цих вакцин вимагає значного часу через тривалий інкубаційний період ВІЛ-інфекції та високу мінливість збудника. Створення високоефективної вакцини проти ВІЛ є актуальною фундаментальною проблемою.