
Мукополісахарідоз 3 типи
Останній перегляд: 23.04.2024

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
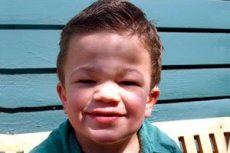
Мукополісахарідоз, тип III (синоніми: синдром Санфіліппо, недостатність лізосомної aN-ацетілглюкозамінідази - мукополисахаридоза Ш А, ацетил-КоА-а-глюкозамінід-N-ацетилтрансферази - мукополисахаридоза III В, N-ацетилглюкозамин-6-сульфатази - мукополісахарідоз III C, сульфамідази - мукополисахаридоза III D).
 [
[Патогенез
До захворювання призводять мутації чотирьох різних генів: лізосомної aN-ацетілглюкозамінідази (мукополісахарідоз III А), ацетил-КоА-а-глюкозамінід-N-ацетилтрансферази (мукополісахарідоз III B), лізосомної N-ацетилглюкозамин-6-сульфатази (мукополісахарідоз III С), сульфамідази (мукополісахарідоз III D). Всі ферменти беруть участь в метаболізмі гепарансульфату.
Ген гепаран-N-сульфатази - SGSH - розташований на довгому плечі хромосоми 17 - 17q25.3. 75,3% відомих на сьогоднішній день мутацій в гені SGSH - точкові. Описано часто зустрічаються мутації, характерні для європейських популяцій - R74C (56% в Польщі і 21% в Німеччині) і R245H (56% в Нідерландах).
Частота мутації R74C становить 47,5%, мутації R245H - 7,5%. Дві інші описані мутації - delll35G і N389S - складають в сукупності 21,7% мутантних алелів.
Ген aN-ацетил-глюкозамінідази - NAGLU - розташований на довгому плечі хромосоми 17 - 17q21. 69% мутацій, знайдених в гені NAGLU, складають міссенс і нонсенс мутації, 26,3% - дрібні делеції і вставки. Ген ацетил-КоА-сс-глюкозамінід-N-ацетилтрансферази - HGSNAT - розташований на короткому плечі хромосоми 8 - 8р11.1. Ген був охарактеризований лише в 2006 р і на сьогоднішній день в ньому виявлено лише кілька мутацій.
Ген N-ацетил-глюкозамін-6-сульфатази - GNS - розташований на довгому плечі хромосоми 12 - 12ql4. У світі зареєстровано 12 пацієнтів з мукополісахаридозом IIID. Описано 4 мутації в гені GNS.
При всіх підтипів мукополисахаридоза III відбувається порушення деградації гепарансульфату, що входить в структуру клітинних мембран, в тому числі мембран нейронів, що корелює з важким нейродегенеративних процесом, обумовленим кортикальной атрофією. Хронічну діарею пояснюють залученістю в патологічний процес вегетативної нервової системи поряд з дисфункцією слизової кишечника. Нейросенсорна туговухість обумовлена, ймовірно, трьома причинами: частими отитами, деформаціями слухових кісточок і аномаліями внутрішнього вуха. Тугоподвижность суглобів - результат деформації метафізів, потовщення суглобової капсули вдруге по відношенню до відкладення в ній гликозаминогликанов і фіброзу. Внутрісіндромальние відмінності в тяжкості захворювання обумовлені виключно залишкової функціональною активністю мутантного ферменту: чим вона вища, тим легше тече захворювання.
Симптоми мукополисахаридоза 3 типи
Клінічний поліморфізм при синдромі Санфіліппо менш виражений, ніж при інших типах мукополисахаридоза. Характерні повільне прогресування захворювання, важкі неврологічні порушення з негрубими симптомами з боку внутрішніх органів і інших систем.
Перші симптоми захворювання зазвичай з'являються у віці від 2-6 років у дітей з попереднім нормальним розвитком. Маніфестних симптоми включають регрес психомоторного і мовного розвитку, психіатричні розлади у вигляді розвитку синдрому гіперактивності, аутичного або агресивної поведінки, порушення сну; діти стають недбалими і неуважними.
Інші часто зустрічаються симптоми - гірсутизм, жорсткі волосся, помірна гепатоспленомегалія, вальгусна деформація кінцівок, коротка шия. Формування грубих рис обличчя за типом гаргоілізм і скелетних деформацій за типом множинного дизостоз слабо виражене при мукополісахарідозов III в порівнянні з іншими типами мукополисахаридоза, що характеризуються Гурлер-фенотипом. Зростання, як правило, відповідає віку, а тугоподвижность суглобів рідко викликає порушення їх функцій. У більшості пацієнтів часто розвиваються остеопороз і остеомаляція. Вторинні скелетні порушення - високий ризик виникнення патологічних переломів. Грубі психоневрологічні розлади найчастіше спостерігають до 6-10-му році життя, вони призводять до вираженої соціальної дезадаптації. Прогресуюча нейросенсорна туговухість властива всім хворим з важкою і помірною формами захворювання. Судоми спостерігають майже у всіх хворих у міру прогресування захворювання.
Перебіг захворювання швидко прогресує, більшість пацієнтів не доживають до 20-річного віку. Вважають, що мукополісахарідоз IIIА - найпоширеніший і важкий тип даного синдрому.
Діагностика мукополисахаридоза 3 типи
Діагноз мукополисахаридоза III підтверджують визначенням рівня екскреції глюкозамігліканов сечі і вимірюванням активності ферментів. У разі мукополисахаридоза III зростає сумарна екскреція глікозаміногліканів в сечі і спостерігається гіперекскреція гепарансульфату. Активність ферментів лізосом, що відповідають певному підтипу мукополисахаридоза III, вимірюють в лейкоцитах або культурі шкірних фібробластів з використанням штучного флюорогенного субстрату.
Пренатальна діагностика можлива шляхом вимірювання активності ферментів в біоптаті ворсин хоріона на 9-11-му тижні вагітності і / або визначення спектру гликозаминогликанов в амніотичної рідини на 20-22-му тижні вагітності. Для сімей з відомим генотипом можливе проведення ДНК-діагностики на ранніх термінах вагітності.
Які аналізи необхідні?
Диференціальна діагностика
Диференціальна діагностика проводиться як всередині групи мукополисахаридозов, так і з іншими лізосомними хворобами накопичення: муколіпідозов, галактосіалідозом, сіалідозом, маннозідозом, фукозідозом, GM1-Гангліозідози.
До кого звернутись?
Лікування мукополисахаридоза 3 типи
На сьогоднішній день ефективних методів терапії для мукополисахаридоза III не розроблено. Показано проведення симптоматичної терапії.
Использованная литература