Медичний експерт статті

Нові публікації
Алкаптонурія - вроджена ферментативна патологія
Останній перегляд: 04.07.2025

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
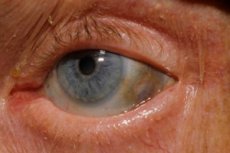
Одним з дуже рідкісних метаболічних розладів є алкаптонурія, що стосується вроджених аномалій метаболізму амінокислоти тирозину.
Цей синдром також може називатися дефіцитом гомогентизатоксидази, гомогентизинурією, спадковим охронозом або хворобою чорної сечі.[ 1 ]
Епідеміологія
Згідно зі статистикою, на 1 мільйон осіб припадає не більше дев'яти випадків алкаптонурії. А в більшості європейських країн – один випадок на 100-250 тисяч живонароджених.
Серед європейських країн винятком є Словаччина (особливо відносно невеликий північно-західний регіон), де поширеність алкаптонурії становить один випадок на 19 000 новонароджених. Найімовірніше, це пов'язано з тим, що серед словацьких ромських сімей, які там проживають, рівень інбридингу (шлюбів між двоюрідними братами та сестрами) є найвищим у Європі: 10-14%. [ 2 ]
Причини алкаптонурії
Точні причини алкаптонурії, як вродженого порушення катаболізму (метаболічного розпаду) ароматичної (гомоциклічної) α-амінокислоти тирозину, встановлені: цей тип метаболічного розладу є наслідком гомозиготних або складних гетерозиготних мутацій одного з тисяч генів на хромосомі 3, точніше, гена HGD у локусі 3q21-q23 на довгому плечі хромосоми. Цей ген кодує нуклеотидні послідовності печінкового ферменту гомогентизат-1,2-діоксигенази [ 3 ] (також званої гомогентизиновою кислотною оксидазою або гомогентизатоксидазою) – залізовмісного металопротеїну, необхідного для одного з етапів розпаду тирозину в організмі. [ 4 ], [ 5 ]
Таким чином, алкаптонурія є дефектом ферменту гомогентизат-1,2-діоксигенази, а точніше, результатом його генетично зумовленого дефіциту або повної відсутності. [ 6 ]
Будучи вродженим дефіцитом ферменту, алкаптонурія успадковується як аутосомно-рецесивна ознака, тобто для виникнення алкаптонурії у дітей обидва батьки повинні мати модифікований ген ферменту, оскільки кожен з них передає дитині лише одну копію гена з двох доступних.
Згідно з останніми даними, існує понад двісті варіантів модифікації гена HGD, причому найчастіше спостерігаються міссенс-мутації, транслокація та сплайсинг.
Фактори ризику
Єдиним фактором ризику розвитку цієї вродженої ензимопатії є її наявність у сімейному анамнезі та успадкування двох модифікованих копій гена HGD, якщо у батьків немає алкаптонурії (ризик передачі аномалії становить 25%), або один з батьків має це захворювання. [ 7 ]
Патогенез
Тирозин відіграє ключову роль у синтезі білків, виробництві хромопротеїнів – пігменту шкіри меланіну, а також гормонів щитовидної залози та катехоламінів-нейромедіаторів.
Механізм регуляції кількості тирозину в клітинах дуже складний, і організм нормалізує його надлишковий вміст шляхом розщеплення. Процес катаболізму тирозину, як і всіх ароматичних амінокислот, є багатоетапним і відбувається в кілька стадій. Кожна стадія метаболічного розщеплення тирозину відбувається за участю певного ферменту та утворенням проміжної сполуки.
Отже, спочатку амінокислота розщеплюється до пара-гідроксифенілпірувату, який перетворюється на алкаптон – 2,5-дигідроксифенілоцтову або гомогентизинову кислоту. Потім алкаптон має перетворитися на малеоцтову кислоту, але цього не відбувається. [ 8 ]
А патогенез алкаптонурії полягає в припиненні біохімічних реакцій катаболізму тирозину на стадії утворення гомогентизинової кислоти: для її розщеплення просто немає необхідного ферменту – гомогентизатоксидази.
Гомогентизинова кислота не використовується організмом і може накопичуватися з виведенням через нирки. Крім того, вона окислюється до бензохіноацетату (бензохінонооцтової кислоти), який, зв'язуючись з молекулами тканин і рідин організму, утворює біополімерні сполуки, забарвлені як меланін.
Накопичення цих проміжних продуктів у тканині призводить до порушення колагенової структури хрящової тканини, що знижує її еластичність – з появою багатьох клінічних ознак алкаптонурії та розвитком ускладнень.
Симптоми алкаптонурії
Алкаптонурія у новонароджених та немовлят характеризується потемнінням сечі. Під впливом повітря сеча на підгузках, пелюшках та спідній білизні стає темно-коричневою; це пов'язано з накопиченням та вивільненням гомогентизинової кислоти, яка окислюється до бензохіноацетату. [ 9 ]
За відсутності інших симптомів алкаптонурія у дітей раннього віку часто не розпізнається своєчасно, оскільки сеча може темніти після кількох годин сечовипускання. За деякими даними, лише п'ята частина дітей віком до 12 місяців, які народилися з дефіцитом цього ферменту, виявляється в клінічних умовах. Тому батькам дуже важливо звертати увагу на догляд за своїми немовлятами.
Крім того, ранні ознаки включають пігментацію (блакитно-сірий колір) склер очей та хрящів вух і носа, що часто називають охронозом.[ 10 ]
З часом з'являються й інші симптоми:
- сильна пігментація шкіри на вилицях, під пахвами та на статевих органах;
- фарбування одягу при контакті з пітними ділянками тіла;
- напади загальної слабкості;
- хрипкий голос.
Слід пам'ятати, що алкаптонурія та охроноз, як зазначалося вище, є синонімами одного й того ж порушення катаболізму тирозину.
Хвороба кленового сиропу та алкаптонурія. Вроджена хвороба кленового сиропу або лейциноз також є порушенням обміну речовин, має той самий тип успадкування, і навіть мутації відбуваються на одній хромосомі, але впливають на ген, що кодує ферментний комплекс α-кетокислот дегідрогенази з розгалуженим ланцюгом. Через це організм не може розщеплювати певні компоненти білків, зокрема, амінокислоти лейцин, ізолейцин та валін. При цьому захворюванні сеча (і вушна сірка) мають солодкуватий запах; крім того, клінічна картина цього типу органічної ацидемії включає гіпопігментацію, коливання артеріального тиску, судоми, блювоту та діарею, зниження рівня глюкози в крові, кетоацидоз, галюцинації тощо. Рівень смертності у дітей досить високий; у дорослих без лікування може настати кома та смерть через набряк мозку.
Альбінізм та алкаптонурію «об’єднує» лише тирозин. Альбінізм, включаючи окулокутанний, спричинений генетичними мутаціями, що впливають на вироблення пігменту меланіну. Вроджені зміни відзначаються в гені TYR на 11-й хромосомі (11q14.3), який кодує тирозиназу – мідьвмісний фермент меланосоми, необхідний для утворення шкірного пігменту на основі продуктів метаболізму тирозину. Це захворювання зустрічається набагато частіше, ніж алкаптонурія.
Ускладнення і наслідки
Спричинені дією проміжних метаболітів тирозину – гомогентизинової та бензохіноноцтової кислот – наслідки та ускладнення алкаптонурії виникають через відкладення реактивних пігментованих полімерів, руйнування колагенових фібрил та погіршення стану хрящів (зі зниженням їх стійкості до механічних навантажень).
З роками, у дорослому віці, розвиваються дегенеративний артрит та остеоартрит великих суглобів (кульшового, крижово-клубового та колінного); міжхребцеві проміжки звужуються (особливо в поперековому та грудному відділах хребта) – з кальцифікацією та утворенням остеофітів; щільність тканини субхондральних кісткових пластинок зменшується, а підлеглі кістки можуть зазнавати патологічного ремоделювання з утворенням наростів та деформації. [ 11 ]
Може спостерігатися пошкодження клапанів серця (аортального та мітрального) та коронарних артерій – з ознаками ішемічної хвороби серця, а також утворення каменів у нирках та передміхуровій залозі – через ту ж кальцифікацію. [ 12 ], [ 13 ]
Діагностика алкаптонурії
Зазвичай діагностика вроджених порушень обміну речовин ґрунтується на дослідженні біологічних рідин організму.
На основі яких аналізів та реакцій можна діагностувати алкаптонурію? Для виявлення гомогентизинової кислоти та визначення її рівня (норма – 20-30 мг на добу, підвищена – 3-8 г) необхідні аналізи сечі. Зразок сечі досліджують за допомогою газової хроматографії або мас-спектрометрії, використовуючи рідинну хроматографію; можливий скринінговий тест на наявність хлориду заліза в сечі. [ 14 ]
Також існує метод експрес-діагностики – визначення алкаптону в засохлих плямах сечі на папері (за інтенсивністю кольору).
При уточненні діагнозу інструментальна діагностика (рентгенографія) передбачає виявлення у пацієнтів рентгенологічних ознак остеоартриту та інших патологій суглобів.
Діагноз підтверджується молекулярно-генетичними методами діагностики спадкових захворювань, такими як генетичне тестування та секвенування ДНК. [ 15 ]
Диференціальна діагностика
Диференціальна діагностика включає гемохроматоз та гостру печінкову недостатність новонароджених, меланінурію, гостру інтермітуючу порфірію, гемофагоцитарний лімфогістіоцитоз, первинну мітохондріальну патологію, ревматоїдний артрит, анкілозуючий спондиліт.
До кого звернутись?
Лікування алкаптонурії
Основним методом лікування алкаптонурії є пероральне застосування великих доз (щонайменше 1000 мг на добу) аскорбінової кислоти. У дітей це збільшує виведення гомогентизинової кислоти з сечею, а у дорослих зменшує вміст у сечі її похідної, бензохінонооцтової кислоти, та уповільнює її зв'язування зі структурами сполучної тканини суглобів та колагену. [ 16 ]
У західноєвропейських клініках тестують препарат Нітизинон (Орфалін) – препарат із групи метаболітів, що пригнічує другий етап катаболізму тирозину: перетворення пара-гідроксифенілпірувату на гомогентизинову кислоту. Однак використання цього фармакологічного засобу призводить до накопичення тирозину та може викликати серйозні побічні ефекти, включаючи помутніння рогівки та світлобоязнь, носові та шлункові кровотечі, печінкову недостатність, зміни в крові тощо. Тим не менш, у Сполучених Штатах Нітизинон схвалений FDA для лікування тирозинемії I типу. [17 ], [ 18 ]
Таким чином, при проблемах із суглобами, спричинених алкаптонурією, проводиться фізіотерапевтичне лікування – ЛФК для збільшення м’язової сили та покращення рухливості суглобів, бальнеотерапія та пелоїдна терапія для обмеження болю.
Хоча тирозин не тільки надходить з їжею, а й виробляється в організмі, пацієнтам з алкаптонурією рекомендується дотримуватися низькобілкової дієти та обмежувати споживання продуктів, багатих на тирозин, насамперед яловичини та свинини, молочних продуктів (особливо сирів), бобових, горіхів та насіння.
Профілактика
Профілактика генних мутацій неможлива, але для запобігання народженню дітей з високим ризиком вроджених вад існує медико-генетичне консультування, яке необхідне перед планованою вагітністю тим парам, у сімейному анамнезі яких є спадкові захворювання. [ 19 ]
Прогноз
Летальні випадки від алкаптонурії трапляються дуже рідко, а смерть може бути спричинена серйозними ускладненнями, що стосуються серця та нирок. Тому загальна тривалість життя людей з алкаптонурією хороша.
Але якість життя знижується через інтенсивний біль у суглобах або хребті зі значним обмеженням рухливості, часто прогресуючий.