Медичний експерт статті

Нові публікації
Синдром Ангельмана у дітей і дорослих
Останній перегляд: 04.07.2025

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.

Існує ряд захворювань, для яких вирази на кшталт «бережи себе і не захворієш» звучать, щонайменше, смішно. Це патології, при яких деякі психічні та фізичні відхилення притаманні організму дитини ще до народження, але батьки в цьому не винні. Такі захворювання викликані мутаціями або аномаліями в хромосомних наборах і називаються хромосомними або генетичними. Синдром Ангельмана, синдром Дауна, синдром Патау, синдром Едвардса, синдром Тернера, синдром Прадера-Віллі – це лише частина генетичних захворювань з досить пристойного списку.
Синдром щасливої людини
Цього разу ми поговоримо про патологію, названу на честь англійського педіатра Гаррі Енджелмана, який вперше порушив питання цієї проблеми у 1965 році, зіткнувшись напередодні у своїй практиці з трьома незвичайними дітьми, об'єднаними спільними своєрідними симптомами. Лікар назвав цих дітей ляльковими дітьми та написав про них статтю, яка спочатку називалася «Діти-маріонетки». Сама стаття та її назва були написані під враженням від картини, побаченої в одному з музеїв Верони. На картині зображено хлопчика, що сміється, і вона називалася «Хлопчик-лялька». Асоціація дитини, зображеної на картині, з трьома дітьми, з якими Енджелман колись зіткнувся у своїй практиці, спонукала педіатра об'єднати дітей в одну групу через хворобу, яку вони мали.
Немає нічого дивного в тому, що згаданих у статті дітей не помітили інші лікарі. Адже на перший погляд здавалося, що у них зовсім інші захворювання, настільки різною була загальна клінічна картина захворювання у 3 різних випадках. Можливо, «нова» хромосомна патологія зацікавила б інших вчених, але на той час генетика ще не була достатньо розвинена, щоб підтвердити гіпотезу англійського лікаря. Тому, після певного інтересу до неї, статтю надовго відклали на задню полицю.
Наступна згадка про синдром Ангельмана, як тепер називалася стаття англійського педіатра Г. Ангельмана, датується початком 80-х років 20 століття. І лише в 1987 році вдалося знайти причину, чому невелика частина дітей народжується з такими відхиленнями, що ззовні вони здаються постійно усміхненими та щасливими. Насправді це зовсім не так, а посмішка – це лише гримаса, за якою ховається нещасна людська душа та біль батьків.
Епідеміологія
Згідно зі статистикою, хромосомна мутація у дитини може розвиватися як на тлі подібних мутацій у батьків, так і за відсутності таких. Чіткої спадкової природи синдрому Ангельмана (СА) немає, але ймовірність розвитку патології у батьків з хромосомними мутаціями досить висока.
Цікаво також, що якщо в сім'ї вже є дитина з синдромом Аспергера, існує одновідсоткова ймовірність народження другої дитини з таким самим розладом, навіть якщо батьки здорові.
Досі немає точної статистики щодо кількості пацієнтів із синдромом Ангельмана. Можливо, причина полягає в різноманітності симптомів, які можуть проявлятися певним складом або взагалі не проявлятися протягом тривалого часу. Вважається, що поширеність захворювання становить: 1 дитина на 20 000 новонароджених. Але ця цифра дуже приблизна.
Причини синдрому Ангельмана
Синдром Ангельмана – це медична назва хромосомної патології, але вона далеко не єдина. Цю хворобу називають синдромом лялькових дітей, синдромом щасливої ляльки, синдромом Петрушки та синдромом сміється ляльки. Люди вигадують усілякі назви (іноді навіть образливі для самих пацієнтів та їхніх батьків), але хвороба є хворобою, як би смішно вона не виглядала і якими б не були причини.
А причини розвитку синдрому Ангельмана, як і багатьох інших генетичних патологій, у всіх випадках полягають у порушеннях у структурі однієї з хромосом або хромосомного набору в цілому. Але в нашому випадку вся проблема полягає в 15-й хромосомі, що передалася від матері. Тобто батьківська хромосома в цьому випадку не має відхилень, а ось жіноча зазнає певних мутацій.
За типом хромосомної аномалії синдром Ангельмана класифікується як хромосомна мутація. Такими мутаціями вважаються:
- Делеція (відсутність ділянки хромосоми, що містить певний набір генів; якщо один з генів відсутній, мова йде про мікроделецію), яка є результатом двох розривів та одного возз'єднання, коли втрачається ділянка вихідної хромосоми.
- Дуплікація (наявність зайвої ділянки в хромосомі, що є копією існуючої), яка в більшості випадків призводить до смерті людини, а рідше до безпліддя.
- Інверсія (розворот однієї з ділянок хромосоми на 180 градусів, тобто у протилежному напрямку, і тоді гени в ній розташовуються у протилежному порядку), коли розірвані кінці хромосоми з'єднуються в порядку, відмінному від початкового.
- Вставка (якщо частина генетичного матеріалу в хромосомі знаходиться поза своїм місцем),
- транслокація (якщо певна ділянка хромосоми прикріплена до іншої хромосоми; така мутація може бути взаємною без втрати ділянок).
Отримуючи мутовану хромосому від нічого не підозрюючої матері, дитина приречена народитися з аномаліями. Найпоширенішою причиною синдрому Ангельмана досі вважається делеція материнської 15-ї хромосоми, коли невелика ділянка відсутня. Менш поширеними мутаціями при синдромі «ляльки, що сміється» вважаються:
- транслокація,
- уніпатернальна дисомія (якщо дитина отримала пару хромосом від батька, материнська хромосома відсутня),
- мутація генів у ДНК, які є одночасно основним будівельним (генетичним) матеріалом та інструкцією для його правильного використання (зокрема, мутація гена ube3a у материнській хромосомі).
Наявність однієї з цих мутацій у батьків є фактором ризику розвитку синдрому Ангельмана у дітей. Але не тільки хромосомні мутації, а й геномні (які пов'язані з кількісною зміною хромосомних наборів і зустрічаються частіше, ніж хромосомні) можуть спровокувати розвиток захворювання у дитини. До поширених геномних мутацій належить хромосомна трисомія (якщо хромосомний набір людини має більше 46 хромосом).
Для появи патології у дитини зовсім не обов'язково, щоб у батьків були хромосомні аномалії. І все ж, є певний відсоток пацієнтів, у яких захворювання є спадковим.
Патогенез
Давайте трохи глибше заглибимося в біологію, а точніше, генетику. Генетична інформація кожного окремого людського організму міститься в 23 парах хромосом. Одна хромосома з пари передається дитині від батька, інша — від матері. Усі пари хромосом відрізняються формою та розміром і несуть певну інформацію. Так, 23-тя пара хромосом (хромосоми X та Y) відповідає за формування статевих ознак дитини (XX — дівчинка, XY — хлопчик, тоді як хромосому Y дитина може отримати лише від батька).
В ідеалі дитина отримує від батьків 46 хромосом, які формують її генетичні характеристики, зумовлюючи її як особистість. Більша кількість хромосом називається трисомією і вважається відхиленням від норми. Наприклад, наявність 47-ї хромосоми в хромосомному наборі (каріотип, що визначає видові та індивідуальні характеристики) спричиняє виникнення синдрому Дауна.
Якщо хромосоми забарвити спеціальним барвником, то під мікроскопом можна побачити смужки різних відтінків вздовж кожної з них. Усередині кожної смужки знаходиться величезна кількість генів. Всі ці смужки пронумеровані вченими та мають фіксоване розташування. Відсутність однієї зі смужок вважається відхиленням від норми. При синдромі Ангельмана дуже часто можна спостерігати відсутність сегментів материнської хромосоми в інтервалі q11-q13, розташованих у довгому плечі, кількість основ ДНК в яких становить лише близько 4 мільйонів.
Основним компонентом хромосоми вважається неймовірно довга молекула ДНК, що містить тисячі генів та десятки й сотні мільйонів азотистих основ. Так, хромосома 15, відповідальна за розвиток синдрому Ангельмана та ряду інших, містить 1200 генів і близько 100 мільйонів основ. Будь-які порушення в структурі молекули ДНК неодмінно вплинуть на зовнішній вигляд і розвиток майбутньої дитини.
Генетична інформація, що міститься в генах, перетворюється на білок або РНК. Цей процес називається експресією генів. Таким чином, генетична інформація, отримана від батьків, отримує як форму, так і зміст, що втілюється в їхньому унікальному спадкоємцю жіночої чи чоловічої статі.
Існує ряд патологій з некласичним типом успадкування, зокрема синдром Ангельмана, при якому гени, отримані від батьків у складі парних хромосом, несуть унікальний відбиток батьків і проявляються по-різному.
Отже, синдром Ангельмана є яскравим прикладом геномного імпринтингу, при якому експресія генів в організмі дитини безпосередньо залежить від того, від якого з батьків були отримані алелі (різні форми одного гена, отримані від батька та матері, розташовані на однакових ділянках парних хромосом). Тобто, до розвитку синдрому призводять лише аномалії в материнській хромосомі, тоді як мутації та структурні порушення батьківської хромосоми викликають зовсім інші патології.
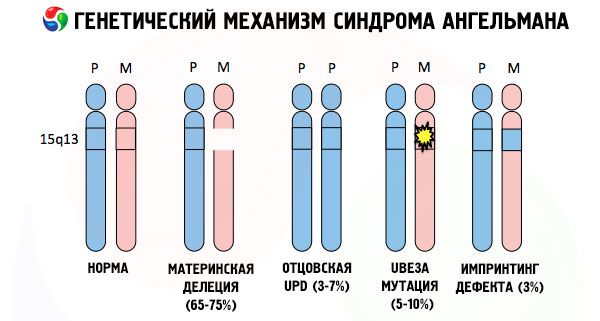
При цій патології спостерігається відсутність певних генів у материнській хромосомі або втрата/зниження активності окремих генів (у переважній більшості випадків гена ube3a, який бере участь у метаболізмі убіквітину – білка, що регулює деградацію інших білків). В результаті у дитини діагностують порушення психічного розвитку та фізичні деформації.
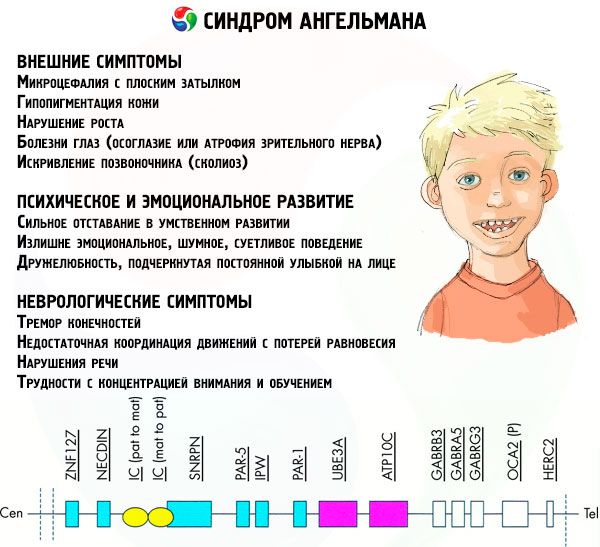
Симптоми синдрому Ангельмана
Симптоми синдрому Ангельмана впливають на різні аспекти життя та розвитку дитини: фізичні, неврологічні, психічні. Виходячи з цього, можна виділити 3 групи симптомів, які свідчать про розвиток цієї патології.
- Зовнішні або фізичні симптоми:
- непропорційно мала голова порівняно з тілом і кінцівками, які мають нормальний розмір,
- занадто широкий рот,
- майже завжди на обличчі (з відкритим ротом) посмішка,
- рідкі зуби,
- вузька верхня губа,
- часто висувається широкий язик,
- виступаюча нижня щелепа,
- загострене підборіддя,
- дуже світла шкіра, часто волосся (альбінізм, пов'язаний з тим, що організм не виробляє пігмент меланін),
- темні плями на світлій шкірі (гіпопігментація через недостатнє вироблення меланіну)
- фізичні або зовнішні симптоми: захворювання очей, такі як косоокість або атрофія зорового нерва,
- викривлення хребта (сколіоз),
- скутість ніг (під час ходьби людина не згинає ноги в колінах через низьку рухливість суглобів, звідси порівняння з ходою ляльки).
- Симптоми, пов'язані з розумовим та емоційним розвитком:
- тяжка розумова відсталість,
- надмірно емоційна, галаслива, метушлива поведінка,
- часті плески в долоні,
- виражена дружелюбність, підкреслена постійною посмішкою на обличчі,
- частий сміх без причини.
- Неврологічні симптоми:
- тремор кінцівок,
- недостатня координація рухів з втратою рівноваги,
- знижений м'язовий тонус,
- різні порушення сну,
- часті істеричні напади в дитинстві,
- порушення мовлення (дитина пізно починає говорити, має погані комунікативні навички та невиразну мову),
- гіперактивність на тлі підвищеної збудливості,
- труднощі з концентрацією уваги та навчанням.
Але це узагальнена картина захворювання. Насправді клінічна картина синдрому Ангельмана значною мірою залежить від стадії розвитку захворювання та типу хромосомної мутації, яка спричинила патологію. Це означає, що симптоми захворювання можуть суттєво відрізнятися у різних пацієнтів, що довгий час не дозволяло відрізнити патологію від інших зі схожою клінічною картиною.
Серед загальної кількості симптомів можна виділити ті, що характерні для всіх пацієнтів без винятку:
- тяжка розумова відсталість,
- неадекватна поведінка (безпричинний сміх, підвищена збудливість, погана концентрація уваги, стан ейфорії),
- недорозвиненість моторних навичок,
- погана координація рухів, атаксія ходи (нерівномірний темп, погойдування з боку в бік тощо), тремор кінцівок.
- порушення розвитку мовлення з переважанням невербальних засобів комунікації.
Серед симптомів, з якими стикається переважна більшість пацієнтів, можна виділити наступні:
- диспропорція між головою та тілом, спричинена затримкою фізичного розвитку,
- у багатьох пацієнтів форма черепа така, що розмір мозку залишається меншим, ніж у здорових людей (мікроцефалія),
- епілептичні напади до 3 років з прогресуючим зниженням сили та частоти у старшому віці,
- спотворення параметрів ЕЕГ (коливання та висока амплітуда низькочастотних хвиль).
Ці симптоми досить поширені, проте у 20% пацієнтів із синдромом Ангельмана їх немає.
Ще рідше вдається діагностувати такі прояви захворювання, як:
- тяжкий або легкий косоокість,
- поганий контроль рухів язика, в результаті чого пацієнти часто висовують язик без причини,
- труднощі з ковтанням і смоктанням, особливо у маленьких дітей,
- порушення пігментації шкіри та очей,
- підняті або зігнуті руки під час ходьби,
- гіперрефлексія,
- розлади сну, особливо в дитячому віці,
- часте слиновиділення,
- невгамовна спрага,
- надмірно активні жувальні рухи,
- гіперчутливість до тепла,
- плоский потилицю,
- виступаюча нижня щелепа,
- гладенькі долоні.
Досить великий відсоток пацієнтів мають проблеми з сечовипусканням, яке вони погано контролюють, порушення дрібної моторики, що створює труднощі в самообслуговуванні та навчанні, а також надмірну вагу. Майже всі пацієнти переживають статеве дозрівання пізніше, ніж здорові однолітки.
Діти із синдромом Ангельмана добре сприймають усне мовлення та розуміють його, але не бажають брати участь у розмові, обмежуючи свою мову кількома десятками слів, необхідних у повсякденному житті. Однак у дорослому віці такі пацієнти виглядають молодшими за своїх однолітків без генетичних патологій.
Багато симптомів синдрому Ангельмана непостійні, тому клінічна картина захворювання значно змінюється з віком. Судоми та епілептичні напади стають рідшими або взагалі зникають, пацієнт стає менш збудливим, покращується сон.
Ускладнення і наслідки
Синдром Ангельмана – це важка, наразі практично невиліковна хромосомна патологія, яка позбавляє пацієнтів можливості жити нормальним життям. Яким буде життя дитини з синдромом Ангельмана, значною мірою залежить від типу хромосомної аномалії.
Дуплікація сегмента хромосоми в більшості випадків несумісна з життям. І навіть якщо такі пацієнти не помирають у немовлячому віці та досягають статевої зрілості, у них немає шансів мати дітей.
Делеція або відсутність частини генів, що найчастіше зустрічається при синдромі Ангельмана, є перешкодою для навчання дитини ходьбі та розмові. У таких дітей спостерігається важча форма розумової відсталості, а епілептичні напади виникають частіше, а їх інтенсивність значно більша, ніж у пацієнтів з іншими хромосомними аномаліями.
Якщо є лише мутація одного гена, за належної уваги та підходу дитину можна навчити основам самообслуговування, спілкування та взаємодії в групі, хоча вона все одно відставатиме від однолітків у розвитку.
Для дітей із синдромом Ангельмана, які за своєю природою добрі, найважливіше – це любов і увага батьків. Тільки в цьому випадку навчання дитини принесе плоди, навіть якщо невеликі. Звичайно, пацієнти з синдромом Ангельмана не зможуть навчатися у звичайній школі. Їм потрібні спеціальні класи, де дітей спочатку навчатимуть концентруватися, а потім поступово дадуть основи шкільних знань.
Діагностика синдрому Ангельмана
Синдром Ангельмана – це вроджена патологія розвитку. Але через певні обставини його часто неможливо діагностувати в немовлячому та ранньому дитинстві. Це пов'язано з неспецифічністю та слабкою вираженістю симптомів у немовлят та дітей віком до 3 років. А поширеність захворювання в нашій країні не настільки велика, щоб лікарі навчилися розпізнавати його серед однолітків.
Синдром Ангельмана у немовлят може проявлятися зниженням м'язового тонусу, що проявляється проблемами з годуванням (слабкість смоктального та ковтального рефлексу), а пізніше труднощами у навчанні ходьбі (такі діти починають ходити набагато пізніше). Ці симптоми є першими ознаками аномалії розвитку у малюка, яка цілком може бути пов'язана з хромосомною аномалією. Тільки генетичний аналіз може підтвердити це припущення.
Особлива увага приділяється дітям, батьки яких мають різні геномні або хромосомні порушення. Адже хвороба може спочатку не проявлятися, і якщо патологію виявити вчасно, почавши інтенсивно працювати з дитиною, можна досягти значно більших успіхів у навчанні, уповільнивши прогресування хвороби.
Якщо у батьків є різні хромосомні аномалії, генетичний аналіз проводиться ще до народження дитини, оскільки СА є однією з патологій, які можна виявити на ембріональній стадії.
Збір матеріалу для генетичних досліджень може здійснюватися двома способами:
- інвазивний (з певним відсотком ризику, оскільки для взяття зразка навколоплідних вод необхідно проникнути в матку),
- неінвазивний (аналіз ДНК дитини з крові матері).
Потім проводяться такі дослідження:
- флуоресцентна гібридизація in situ (метод FISH) – зв'язування ДНК-зонда, міченого спеціальним барвником, з досліджуваною ДНК з подальшим дослідженням під мікроскопом.
- аналіз мутацій у гені ube3a та імпринтованих генах,
- Аналіз метилювання ДНК за допомогою спеціальних методів, що використовуються в генетиці.
Генетичні тести дають досить точну інформацію у випадку хромосомних аномалій, а це означає, що майбутні батьки заздалегідь знають, до чого готуватися. Однак є винятки. У певної групи пацієнтів за наявності всіх симптомів, що вказують на патологію, результати тестів залишаються нормальними. Тобто виявити патологію можна лише уважно спостерігаючи за дитиною з раннього дитинства: як вона харчується, коли почала ходити та говорити, чи згинає ноги під час ходьби тощо.
Окрім методу FISH, серед інструментальних методів діагностики синдрому Ангельмана можна виділити томографію (КТ або МРТ), яка допомагає визначити стан і розміри мозку, та електроенцефалограму (ЕЕГ), яка показує, як працюють окремі відділи мозку.
Лікарі зазвичай ставлять остаточний діагноз у віці 3-7 років, коли у пацієнта вже є більшість симптомів і видно динаміку розвитку захворювання.
Які аналізи необхідні?
Диференціальна діагностика
Синдром Ангельмана – це генетична патологія, яка практично не має специфічних проявів. Більшість симптомів можуть однаково свідчити як про АС, так і про інші генетичні патології.
Диференціальна діагностика синдрому Ангельмана проводиться з такими патологіями:
- Синдром Пітта-Хопкінса (хворі характеризуються розумовою відсталістю, життєрадісним характером, посмішкою, у них досить великий і широкий рот, відзначається мікроцефалія). Відмінністю є напади гіпервентиляції та затримка дихання у стані неспання.
- Синдром Крістіансона (хворі – розумово відсталі люди з життєрадісною вдачею, не здатні говорити, характеризуються мікроцефалією, атаксією, судомами, мимовільними рухами м’язів).
- Синдром Мовата-Вільсона (симптоми: розумова відсталість, епілептичні напади, загострене підборіддя, відкритий рот, щасливий вираз обличчя, мікроцефалія). Відмінні риси: велика відстань між очима, очі розкосі всередину, закруглений кінчик носа, загнута назад вушна раковина.
- Синдром Кабукі (характеризується легкою та помірною розумовою відсталістю, проблемами з мовленням та руховими функціями, м’язовою слабкістю, епілептичними нападами, мікроцефалією, довгими інтервалами між свербіннями та порушенням координації). Характеризується дугоподібними бровами, вивернутою латеральною частиною нижньої повіки, широко розставленими очима, довгими очними щілинами з довгими, густими віями.
- Синдром Ретта (диференціація від АС у жінок). Симптоми: затримка розвитку мовлення, судоми, мікроцефалія. Відмінність полягає в тому, що на обличчі відсутній радісний вираз, спостерігаються напади апное та апраксії, що прогресує з часом.
- Аутосомно-рецесивний синдром затримки психічного розвитку 38 (симптоми: виражена розумова відсталість із затримками розвитку моторних навичок та мовлення, м'язова слабкість, проблеми з годуванням у немовлячому віці, імпульсивність). Відмінною рисою є блакитний колір райдужної оболонки ока.
- Синдром дуплікації гена MECP 2 (диференціація від SA у чоловіків). Симптоми: тяжка розумова відсталість, м'язова слабкість з дитинства, проблеми з мовленням або його відсутність, епілепсія. Відмінні ознаки: прогресуюча міопатія, постійно рецидивуючі інфекції.
- Синдром Кліфстри (симптоми: проблеми з мовленням та мисленням, м'язова слабкість, порушення сну, неуважність, відкритий рот, гіперактивність, судоми, атаксія, порушення рівноваги). Відмінні риси: плоске обличчя, короткий кирпатий ніс, широко розставлені очі, велика вивернута нижня губа, агресивні спалахи.
- Синдром Сміта-Магеніса (характеризується судомами, проблемами зі сном, порушеннями інтелектуального та моторного розвитку). Відмінними рисами є широке та плоске обличчя й опуклий лоб.
- Синдром Кулена-де Вріса (легка та помірна розумова відсталість, м'язова слабкість, судоми, дружелюбність). Відмінні риси: довге обличчя з високим чолом, виступаючі вуха, розкосі очі, висока рухливість суглобів, вроджені вади серця.
- Синдром Фелана-Макдерміда (симптоми: розумова відсталість, порушення мовлення або відсутність мовлення). Відмінні риси: великі кисті рук з розвиненими м'язами, м'язова слабкість від народження, слабке потовиділення.
Такі патології, як дефіцит аденілсукцинату, аутосомно-рецесивний синдром розумової відсталості 1 типу, синдром дуплікації хромосоми 2q23.1, синдроми гаплонедостатності генів FOXG1, STXBP1 або MEF2C та деякі інші, можуть «похвалитися» симптомами, подібними до синдрому Ангельмана.
Завдання лікаря — поставити точний діагноз, диференціюючи синдром Ангельмана від патологій зі схожими симптомами, та призначити ефективне лікування, що відповідає діагностованій стадії захворювання.
Лікування синдрому Ангельмана
Синдром Ангельмана – одна з тих патологій, для яких медицина все ще шукає ефективне лікування. Етіологічне лікування захворювання перебуває на стадії розробки різних методів і засобів, багато з яких ще не випробувані на людях. Це означає, що поки що лікарям доводиться обмежуватися симптоматичною терапією, яка допомагає хоч якось полегшити незавидне становище дітей і дорослих із синдромом маріонетки, які страждають від епілептичних нападів, слинотечі, гіпотензії та розладів сну.
Таким чином, зменшити частоту та силу епілептичних нападів можна за допомогою правильно підібраного протисудомного препарату. Але вся складність полягає в тому, що напади у пацієнтів із СА відрізняються від звичайних епілептичних нападів тим, що для них характерні кілька типів нападів, а це означає, що стан можна полегшити, вводячи одразу кілька препаратів.
Найпопулярнішими протисудомними засобами, що використовуються для лікування синдрому Ангельмана, є: вальпроєва кислота, топірамат, ламотриджин, леветирацетам, клоназепам та препарати на їх основі. Рідше використовуються препарати на основі кармазепіну, фенітоїну, фенобарбіталу, етосуксиміду, оскільки деякі з них можуть провокувати парадоксальний ефект, що полягає в посиленні та збільшенні частоти епілептичних нападів. Це трапляється, якщо препарат використовується в складі монотерапії.
Для лікування слинотечі зазвичай використовуються два методи: медикаментозний (препарати, що пригнічують вироблення слини) та хірургічний, який передбачає реімплантацію слинних проток. Але у випадку сальмональної анемії ці методи вважаються неефективними, і питання залишається відкритим. Батькам та тим, хто доглядає за такими пацієнтами, доводиться звертати особливу увагу на це питання, оскільки самі пацієнти зазвичай не контролюють слинотечу, а деякі просто не в змозі самостійно про себе подбати.
Ще однією проблемою є коротка тривалість сну. Часто діти із синдромом Ангельмана сплять не більше 5 годин, що негативно впливає на функціонування всього організму. Легко збудливі, активні діти, які люблять ігри та спілкування (навіть якщо намагаються обмежити себе невербальними методами), помітно втомлюються протягом дня. Щоб добре відпочити, організму потрібен глибокий, повноцінний сон, але саме в цьому і полягає заковика.
Здавалося б, седативних препаратів (фенотіазинів та атипових антипсихотиків), що заспокоюють нервову систему, має бути достатньо для покращення сну у збудливих пацієнтів. Але у випадку з АС застосування таких препаратів загрожує виникненням негативних ефектів. Тому лікарі все ж таки віддають перевагу м’яким снодійним, таким як Мелатонін (натуральний гормональний препарат на основі гормону сну), який дають пацієнтам за годину до сну в кількості 1 таблетки, та Дифенгідрамін. Кратність прийому та дозування якого визначає лікар залежно від стану та віку пацієнта.
Іноді у пацієнтів із синдромом Ангельмана виникають проблеми з травленням та стільцем. Поліпшити стілець можна за допомогою проносних засобів (бажано рослинних).
Або ж можна підійти до проблеми інакше, як це зробили американські лікарі, спираючись на деякі методи лікування аутизму, адже багато симптомів, характерних для АС, також характерні для аутизму (імпульсивність, мимовільні рухи, повторювані дії, дефіцит уваги, проблеми з комунікацією тощо). Було відзначено, що введення гормону секретину, який нормалізує травлення та стілець, позитивно впливає на увагу пацієнтів, а окситоцин сприяє покращенню когнітивних здібностей та пам'яті дитини, корекції поведінки.
Правда, одних лише гормонів недостатньо, особливо коли йдеться про дітей. При синдромі Ангельмана показана поведінкова терапія, робота з психологом та логопедом (навчання методам невербального спілкування та мові жестів). Навчання таких дітей має базуватися на індивідуальній програмі за участю спеціально навчених педагогів, психолога та батьків. На жаль, це можливо не скрізь, і сім'ї залишаються наодинці зі своєю проблемою.
Оскільки багато молодих пацієнтів з АС страждають від низького м’язового тонусу та проблем із суглобами, велика увага приділяється фізіотерапії. Найчастіше лікарі вдаються до використання парафінових аплікацій, електрофорезу та магнітотерапії.
Активний тонізуючий масаж та спеціальні вправи лікувальної фізкультури допоможуть хворій дитині через деякий час стати на ноги та впевнено ходити. Особливо корисна в цьому плані аквагімнастика, яку рекомендують при СА в прохолодній воді. Вона підвищує м’язовий тонус і вчить дитину контролювати своє тіло та координувати рухи.
Протисудомне лікування
Найнебезпечнішим симптомом синдрому Ангельмана є судоми, подібні до епілептичних. Цей симптом спостерігається у 80% пацієнтів, а це означає, що всім їм необхідно призначити ефективне протисудомне лікування.
Лікування епілептичних нападів проводиться за допомогою вітамінів та протисудомних препаратів. При синдромі Ангельмана, що супроводжується судомним синдромом, корисними будуть вітаміни групи В, а також вітаміни С, D та Е. Але самостійне призначення вітамінотерапії в цьому випадку дуже небезпечне, оскільки безконтрольний прийом вітамінів може знизити ефективність протиепілептичних препаратів та спровокувати нові, більш сильні та тривалі напади.
Підбір протисудомних препаратів та призначення їх ефективного дозування також повинен здійснювати лікар-спеціаліст. Він також вирішує, чи буде достатньо одного препарату, чи пацієнту доведеться приймати 2 або більше препаратів протягом тривалого часу.
Більшості пацієнтів лікарі призначають препарати вальпроєвої кислоти (Вальпроєва кислота, Депакін, Конвулекс, Вальпарин тощо), які запобігають судомам та покращують настрій і психічний стан пацієнтів.
Вальпроєва кислота випускається у формі таблеток, сиропу та розчинів для ін'єкцій. Найпопулярнішим препаратом є препарат пролонгованої дії «Депакін» у таблетках та у вигляді розчину для внутрішньовенного введення. Дозування препарату визначається лікарем індивідуально залежно від ваги, віку та стану пацієнта.
Препарат приймають під час їжі 2 - 3 рази на день. Середня добова доза становить 20-30 мг на 1 кілограм ваги пацієнта, максимальна - 50 мг/кг на добу.
Протипоказання до застосування. Не застосовувати при порушеннях функції печінки та підшлункової залози, геморагічному діатезі, гепатиті, порфірії та підвищеній чутливості до препарату.
Побічні ефекти включають тремор рук, розлади травлення та стільця, а також зміни маси тіла.
«Топірамат» також є препаратом вибору при СА. Він випускається у формі таблеток і використовується як у складі монотерапії, так і в комбінації з іншими препаратами.
Спосіб застосування та дозування. Приймати таблетки перорально незалежно від прийому їжі. Початкова добова доза для дорослих становить 25-50 мг, для дітей – 0,5-1 мг/кг. Щотижня дозування збільшують за призначенням лікаря.
Препарат не слід приймати під час вагітності та лактації, а також при підвищеній чутливості до його компонентів. Препарат має багато різних побічних ефектів.
Препарати, які лікар може призначити при синдромі Ангельмана: Кломазепам, Рівотрил, Ламотриджин, Сейзар, Ламіктал, Леветирацетам, Кеппра, Епітерра тощо.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Традиційна медицина та гомеопатія
Традиційна медицина, як і гомеопатичні препарати, звичайно, відносно безпечна, але ефективність такого лікування синдрому Ангельмана можна вважати суперечливою.
Хоча народне лікування все ж може допомогти в деяких речах. Йдеться про купірування епілептичних нападів. У цьому плані лікування травами може бути досить ефективним.
Гарний ефект дає лікувальний збір на основі півонії, солодки та ряски (компоненти беруться в рівних кількостях). Трави потрібно перемолоти на борошно. Через 2 тижні від початку прийому можна помітити значне зниження частоти нападів.
Також корисний при спазмах відвар лаванди (1 чайна ложка на склянку окропу). Суміш кип’ятять 5 хвилин і настоюють півгодини. Ліки приймають на ніч протягом 14 днів.
Водний (або спиртовий) настій пустирника вважається ефективним при епілептичних нападах.
З гомеопатичних препаратів для профілактики судом при синдромі Ангельмана можна використовувати ліки на основі ромашки та пустирника, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Але слід враховувати, що тільки лікар-гомеопат може призначити ефективні та безпечні дози препаратів у кожному конкретному випадку.
Профілактика
Як читач, мабуть, вже зрозумів, медицина поки що не в змозі запобігти генним мутаціям та іншим хромосомним аномаліям, втім, як і виправити ситуацію. Це може статися з будь-ким, адже діти із синдромом Ангельмана народжуються у здорових батьків, і генетика, яка наразі є однією з найменш вивчених галузей медицини, поки що не може цього пояснити.
Єдине, що можна зробити, це відповідально підійти до планування вагітності, вчасно стати на облік та пройти обстеження. Але знову ж таки, такий захід буде радше освітнім, ніж профілактичним, як і будь-які обстеження. Але молоді батьки заздалегідь знатимуть, до чого готуватися, і в разі позитивної відповіді вирішуватимуть, чи можуть вони взяти на себе таку відповідальність, як виховання хворої дитини.
Прогноз
Прогноз при синдромі Ангельмана залежить від характеру хромосомної аномалії та своєчасності її виявлення. Найбільше страждають ті діти, чия 15-та хромосома містить «прогалини» в генах (делеції). Ймовірність того, що такі пацієнти почнуть ходити та говорити, вкрай низька. Інші випадки можна виправити за допомогою ретельного підходу та любові до своєї дитини.
На жаль, такі пацієнти не зможуть стати повноцінними членами суспільства, незважаючи на те, що вони далеко не дурні, розуміють мову та її значення. Однак у них будуть проблеми зі спілкуванням до кінця життя. Пацієнтів можна навчати жестовій мові з дитинства, але не можна змусити спілкуватися за допомогою слів. Словниковий запас «говорящих» пацієнтів обмежений мінімумом слів, що використовуються в повсякденному житті (5-15 слів).
Що стосується тривалості життя та загального стану здоров'я пацієнтів із синдромом Ангельмана, то тут показники коливаються навколо середніх значень. У дорослому віці пацієнти здебільшого стикаються з такими проблемами зі здоров'ям, як сколіоз та ожиріння, які за правильного підходу до лікування не загрожують життю.