Медичний експерт статті

Нові публікації
Синдром Трічер Коллінза
Останній перегляд: 04.07.2025

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.

Внутрішньоутробні порушення процесів розвитку кісток викликають серйозні краніофаціальні деформації, і одним з різновидів такої патології є синдром Трічера Коллінза (СТК) або мандибулофасціальний, тобто щелепно-лицьовий дизостоз.
Код захворювання за МКХ 10: клас XVII (вроджені аномалії, деформації та хромосомні порушення), Q75.4 - дизостоз нижньощелепної області.
Причини синдрому Трічер Коллінза
Цей синдром був названий на честь видатного британського офтальмолога Едварда Трішера Коллінза, який описав основні риси патології понад сто років тому. Однак європейські лікарі частіше називають цей тип аномалії лицьових та щелепних кісток хворобою або синдромом Франческотті – на основі масштабних досліджень швейцарського офтальмолога Адольфа Франческотті, який у середині минулого століття ввів термін «мандибулофасціальний дизостоз». У медичних колах також використовується назва синдром Франческотті-Коллінза.
Синдром Трішера Коллінза спричинений мутаціями в гені TCOF1 (у локусі хромосоми 5q31.3-33.3), який кодує ядерцевий фосфопротеїн, відповідальний за формування краніофаціальної частини ембріона людини. Внаслідок передчасного зменшення кількості цього білка порушується біогенез та функції рРНК. За даними генетиків з програми дослідження геному людини, ці процеси призводять до зменшення проліферації ембріональних клітин нервового гребеня – гребеня вздовж нервової борозни, який під час ембріонального розвитку замикається в нервову трубку.
Формування тканин обличчя відбувається завдяки трансформації та диференціації клітин верхньої (головної) частини нервового гребеня, які мігрують по нервовій трубці до області першої та другої зябрових дуг ембріона. А дефіцит цих клітин викликає краніофаціальні деформації. Критичний період для виникнення аномалій – з 18 по 28 день після запліднення. Після завершення міграції клітин нервового гребеня (на четвертому тижні вагітності) утворюються майже всі пухкі мезенхімальні тканини в області обличчя, які пізніше (з 5 до 8 тижнів) диференціюються в скелетні та сполучні тканини всіх частин обличчя, шиї, гортані, вуха (включаючи внутрішнє вухо) та майбутніх зубів.
Патогенез
Патогенез синдрому Трічера Коллінза часто є сімейним, і аномалія успадковується за аутосомно-домінантним типом, хоча трапляються випадки аутосомно-рецесивної передачі дефекту (з мутаціями в інших генах, зокрема, POLR1C та POLR1D). Найбільш непередбачуваним щодо щелепно-лицьового дизостозу є те, що мутація успадковується дітьми лише у 40-48% випадків. Тобто у 52-60% пацієнтів причини синдрому Трічера Коллінза не пов'язані з наявністю аномалії в родині, і вважається, що патологія виникає в результаті спорадичних генних мутацій de novo. Найімовірніше, нові мутації є наслідками тератогенного впливу на плід під час вагітності.
Серед тератогенних причин цього синдрому фахівці називають великі дози етанолу (етилового спирту), радіацію, сигаретний дим, цитомегавірус та токсоплазму, а також гербіциди на основі гліфосату (Раундал, Гліфор, Торнадо тощо). А до списку ятрогенних факторів входять препарати від акне та себореї з 13-цис-ретиноєвою кислотою (Ізотретиноїн, Аккутан); протисудомний препарат Фенітоїн (Дилантин, Епанутин); психотропні препарати Діазепам, Валіум, Реланіум, Седуксен.
Симптоми синдрому Трічер Коллінза
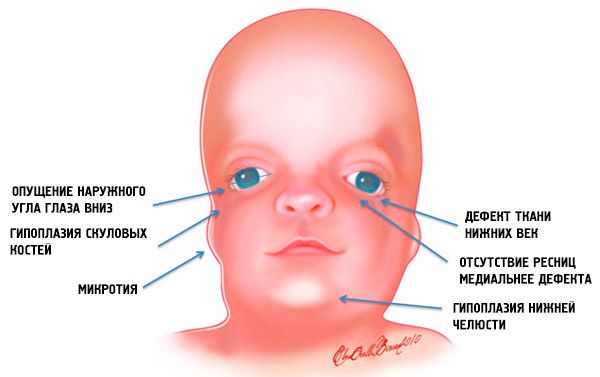
Здебільшого клінічні ознаки мандибулофасціального дизостозу та ступінь їх вираженості залежать від особливостей прояву генних мутацій. І перші ознаки цієї аномалії в більшості випадків видно у дитини одразу після народження: обличчя при синдромі Трічера Коллінза має характерний вигляд. Причому морфологічні аномалії зазвичай двосторонні та симетричні.
Найбільш очевидними симптомами синдрому Трішера Коллінза є:
- недорозвинення (гіпоплазія) лицьових кісток черепа: виличної кістки, виличкових відростків лобової кістки, бічних крилоподібних пластинок, навколоносових пазух, нижньої щелепи та випинань кісткових епіфізів (виростків);
- недорозвинення кісток нижньої щелепи (мікрогнатія) та більш тупий, ніж зазвичай, кут нижньої щелепи;
- ніс нормального розміру, але здається великим через гіпоплазію надбрівних дуг та недорозвинення або відсутність виличних дуг у скроневій ділянці;
- очні щілини спрямовані вниз, тобто форма очей аномальна, із зовнішніми куточками, опущеними вниз;
- дефекти нижніх повік (колобома) та часткова відсутність вій на них;
- неправильної форми вушні раковини з широким діапазоном відхилень, включаючи їх розташування в кутку нижньої щелепи, відсутність мочок, сліпі фістули між козелком вуха та кутом рота тощо;
- звуження або закриття (атрезія) зовнішнього слухового проходу та аномалії розвитку кісточок середнього вуха;
- відсутність або гіпоплазія привушних слинних залоз;
- гіпоплазія глотки (звуження глотки та дихальних шляхів);
- незрощення твердого піднебіння (щілина піднебіння), а також відсутність, вкорочення або нерухомість м'якого піднебіння.
Такі анатомічні аномалії у всіх випадках мають ускладнення. Це функціональні порушення слуху у вигляді кондуктивної втрати слуху або повної глухоти; порушення зору через неправильне формування очних яблук; дефекти піднебіння викликають труднощі з годуванням та ковтанням. Існують порушення зубного прикусу (мальокклюзія), пов'язані з дефектами щелепи, що, у свою чергу, викликає проблеми з жуванням та артикуляцією. Патології м'якого піднебіння пояснюють носовий голос.
Ускладнення і наслідки
Наслідки щелепно-лицьових аномалій при синдромі Трішера Коллінза полягають у тому, що при народженні інтелектуальні здібності дитини нормальні, але через дефекти слуху та інші порушення спостерігається вторинна розумова відсталість.
Крім того, діти з такими вадами гостро відчувають свою неповноцінність і страждають, що негативно впливає на їхню нервову систему та психіку.
Діагностика синдрому Трічер Коллінза
Постнатальна діагностика синдрому Трішера Коллінза по суті базується на клінічних ознаках. Краніофаціальний дизостоз легко ідентифікується, коли синдром повністю виражений, але коли присутні мінімально виражені симптоми патології, можуть виникнути проблеми з встановленням правильного діагнозу.
У цьому випадку особливу увагу слід приділити оцінці всіх функцій, пов'язаних з аномаліями, особливо тих, що впливають на дихання (через ризик апное сну). Також слід оцінювати та контролювати ефективність годування та насичення гемоглобіну киснем.
Пізніше, на 5-6-й день після народження, ступінь пошкодження слуху потрібно буде визначити за допомогою аудіологічного тестування, яке слід проводити в пологовому будинку.
Призначається обстеження, під час якого проводиться інструментальна діагностика шляхом флюороскопії краніофаціальної дисморфології; пантомографії (панорамної рентгенографії кісткових структур лицевого черепа); повної краніальної комп'ютерної томографії в різних проекціях; КТ або МРТ головного мозку для визначення стану внутрішнього слухового проходу.
Найраніша – пренатальна – діагностика щелепно-лицьових аномалій за наявності синдрому Трішера Коллінза в сімейному анамнезі можлива шляхом біопсії ворсин хоріона на 10-11 тижні вагітності (процедура загрожує викиднем та інфікуванням матки).
Також беруть аналізи крові у членів сім'ї; на 16-17 тижні вагітності аналізують навколоплідні води (трансабдомінальний амніоцентез); на 18-20 тижні вагітності проводять фетоскопію та беруть кров із судин плода плаценти.
Але найчастіше УЗД використовується в пренатальній діагностиці цього синдрому у плода (на 20-24 тижні вагітності).
Які аналізи необхідні?
Диференціальна діагностика
Ці ж методи використовуються фахівцями, коли потрібна диференціальна діагностика для розпізнавання легкого синдрому Трішера-Коллінза та диференціації його від інших вроджених аномалій черепно-лицьових кісток, зокрема: синдромів Аперта, Крузона, Нагера, Петерса-Хьюелса, Хеллермана-Стефа, а також геміфаціальної мікросомії (синдрому Голденхара), гіпертелоризму, передчасного зрощення черепних швів (краніосиностозу) або порушення зрощення кісток обличчя (краніосиностозу).
Лікування синдрому Трічер Коллінза
Як і у всіх випадках генетично зумовлених вроджених дефектів, лікування важких форм синдрому Трічера Коллінза є виключно паліативним, оскільки терапевтичних методів для таких патологій просто немає. Спектр і ступінь деформацій при цьому синдромі є широкими і, отже, характер та інтенсивність медичного втручання також мають багато варіантів.
Для корекції та покращення слуху використовуються слухові апарати, а для покращення мовлення – сеанси логопедичної терапії.
Хірургічні втручання потрібні в ранньому віці у важких випадках звуження дихальних шляхів (виконується трахеостомія) та гортані (для годування проводиться гастростомія). Також може знадобитися хірургічна корекція піднебіння.
Операції з подовження нижньої щелепи виконуються у віці 2-3 років або пізніше. Реконструкція м’яких тканин включає корекцію колобоми нижньої повіки та пластику вушних раковин.
Прогноз
Який прогноз при цій патології? Він залежить від ступеня деформації та інтенсивності симптомів. Синдром Трічера Коллінза є довічним діагнозом.
 [ 25 ]
[ 25 ]