
Алкаптонурія – вроджена ферментативна патологія
Останній перегляд: 23.04.2024

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
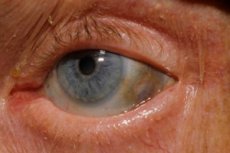
Одне з дуже рідкісних порушень обміну речовин - алкаптонурія - відноситься до вроджених аномалій метаболізму амінокислоти тирозину.
Також даний синдром може називатися дефіцитом гомогентізатоксідази, гомогентізінуріей, спадковим охроноз або хворобою чорної сечі. [1]
Епідеміологія
За статистикою, на 1 млн. Населення припадає не більше дев'яти випадків алкаптонуріі. А в більшості країн Європи - один випадок на 100-250 тис. Живонароджених немовлят.
Серед європейських країн винятком є Словаччина (особливого відносно невеликий по території північно-західний регіон), де поширеність алкаптонуріі становить один випадок на 19 тис. Новонароджених. Цілком ймовірно, це пов'язано з тим, що серед проживаючих там сімей словацьких ромів рівень інбридингу (шлюбів між двоюрідними родичами) найвищий в Європі: 10-14%. [2]
Причини алкаптонуріі
Точні причини алкаптонуріі, як вродженого розладу катаболізму (метаболічного розпаду) ароматичної (гомоцікліческой) α-амінокислоти тирозину, встановлені: даний тип порушення обмінних процесів є наслідком гомозиготних або складних гетерозиготних мутацій одного з тисячі генів на хромосомі 3, точніше - гена HGD в локусі 3q21 -q23 на довгому плечі хромосоми. Даним геном кодуються нуклеотидні послідовності ферменту печінки гомогентізат-1,2-діоксигенази [3](також званого оксидазой гомогентизиновой кислоти або гомогентізатоксідазой) - залізовмісного металопротеїни, необхідного для одного з етапів розщеплення тирозину в організмі. [4], [5]
Таким чином, алкаптонурія - дефект ферменту гомогентізат-1,2-діоксигенази, точніше, результат його генетично зумовленого дефіциту або повної відсутності. [6]
Представляючи собою вроджену ферментопатія, алкаптонурія успадковується як аутосомний рецесивний ознака, тобто, щоб виникла алкаптонурія у дітей, повинен бути видозмінений ген зазначеного ферменту в обох батьків, оскільки кожен з них передає дитині лише одну копію гена з наявних двох.
За останніми даними, налічується понад двісті варіантів видозміни гена HGD, і найчастіше спостерігаються міссенс-мутації, транслокація і сплайсинг.
Фактори ризику
Єдиний фактор ризику розвитку даної вродженої ферментопатії - наявність її в сімейному анамнезі і успадкування двох видозмінених копій гена HGD, якщо у батьків алкаптонурія не проявляється (ризик передачі аномалії - 25%), або один з батьків має цей розлад. [7]
Патогенез
Найважливішу роль тирозин відіграє в синтезі протеїнів, виробленні хромопротеїни - шкірного пігменту меланіну, а також тиреоїдних гормонів і катехоламінових нейротрансмітерів.
Механізм регулювання кількості тирозину в клітинах дуже складний, і його надмірний вміст організм нормалізує шляхом розщеплення. Процес катаболізму тирозину, як і всіх ароматичних амінокислот, багатоступінчастий і протікає в кілька стадій. При цьому кожен етап метаболічного розпаду тирозину проходить за участю певного ферменту і утворенням проміжного з'єднання.
Так, спочатку амінокислота розщеплюється до пара-гідроксіфенілпірувата, який перетворюється в алкаптон - 2,5-дігідроксіфенілуксусную або гомогентизинової кислоту. Далі алкаптон повинен трансформуватися в малеуксусную кислоту, але цього не відбувається. [8]
І патогенез алкаптонуріі полягає в зупинці біохімічних реакцій катаболізму тирозину на етапі утворення гомогентизиновой кислоти: для її розщеплення просто немає потрібного ферменту - гомогентізатоксідази.
Гомогентізіновая кислота організмом не використовується і може накопичуватися з виведенням через нирки. Крім того, відбувається її окислення до бензохіноацетата (бензохінонуксусной кислоти), який, зв'язуючись з молекулами тканин і рідин організму, утворює біополімерні з'єднання, пофарбовані подібно меланину.
Накопичення в тканини даних проміжних продуктів призводить до порушення структури колагену хрящової тканини, через що знижується її еластичність - з появою багатьох клінічних ознак алкаптонуріі і розвитком ускладнень.
Симптоми алкаптонуріі
Алкаптонурия у новонароджених і дітей грудного віку проявляється потемніння сечі. При контакті з повітрям колір сечі на підгузниках, пелюшках і білизна стає темно-коричневим; це пов'язано з накопиченням і виділенням гомогентизиновой кислоти, яка окислюється до бензохіноацетата. [9]
При відсутності інших симптомів нерідко алкаптонурія у дітей в ранньому віці своєчасно не розпізнається, так як після сечовипускання сеча може потемніти через кілька годин. За деякими даними, в умовах клінік виявляється лише п'ята частина дітей до 12 місяців, які народилися з даної ферментопатією. Тому дуже важливо увагу батьків до догляду за немовлям.
Крім того, перші ознаки включають пігментацію (синювато-сірого кольору) очних склер і хрящів вушних раковин і носа, яку часто називають охроноз. [10]
Згодом з'являються й інші симптоми:
- сильна пігментація шкіри на вилицях, пахвових западин і геніталій;
- фарбування одягу при контакті з потіють ділянками тіла;
- напади загальної слабкості;
- хрипкий голос.
Слід мати на увазі, що алкаптонурія і охроноз , як уже зазначалося вище, є синонімічні назвами одного і того ж порушення катаболізму тирозину.
Хвороба кленового сиропу і алкаптонурія. Вроджена хвороба кленового сиропу або лейциноз теж відноситься до метаболічних порушень, має той же тип спадкування, і навіть мутації відбуваються на тій же хромосомі, але стосуються гена, що кодує ферментний комплекс дегідрогенази розгалужених α-кетокислот. Через це організм не може розщеплювати певні компоненти білків, зокрема, амінокислоти лейцин, ізолейцин і валін. При цьому захворюванні сеча (і вушна сірка) має солодкий запах; крім того, в клінічній картині цього типу органічної ацидемії спостерігаються гипопигментация, коливання артеріального тиску, судоми, блювота і діарея, падіння рівня глюкози в крові, кетоацидоз, галюцинації і ін. Рівень смертності у дітей досить високий; у дорослих без лікування може наступити кома і летальний результат внаслідок набряку мозку.
Альбінізм і алкаптонурія «об'єднує» тільки тирозин. Альбінізм , в тому числі і окулокутанний, викликається генетичними мутаціями, які впливають на вироблення пігменту меланіну. Вроджені зміни відзначаються в гені TYR на хромосомі 11 (11q14.3), який кодує тирозиназу - містить мідь фермент меланосом, необхідний для утворення шкірного пігменту на основі продуктів метаболізму тирозину. Дане захворювання набагато більш поширене, ніж алкаптонурія.
Ускладнення і наслідки
Зумовлені дією проміжних метаболітів тирозину - гомогентизиновой і бензохінонуксусной кислот - наслідки і ускладнення алкаптонуріі з'являються через осадження реактивних пігментованих полімерів, руйнування фібрил колагену і погіршення стану хрящів (зі зниженням їх стійкості до механічних навантажень).
Через роки, в зрілому віці, розвивається дегенеративний артрит і остеоартрит великих суглобів (тазостегнових, крижово-клубових і колінних); звужуються міжхребцеві простору (особливо поперекового і грудного відділів хребта) - з кальцификацией і утворенням остеофитов; знижується щільність тканини субхондральних кісткових пластин, а підлягають кістки можуть піддаватися патологічного ремоделювання з утворенням наростів і деформацією. [11]
Може спостерігатися пошкодження серцевих клапанів (аортальних і мітральних) і коронарних артерій - з ознаками ішемічної хвороби серця, а також утворення каменів в нирках і передміхуровій залозі - внаслідок того ж кальциноза. [12], [13]
Діагностика алкаптонуріі
Зазвичай діагностика вроджених порушень обміну речовин заснована на дослідженні біологічних рідин організму.
На основі яких аналізів і якими реакціями можна діагностувати Алкаптонурія? Необхідні аналізи сечі - для в виявлення гомогентизиновой кислоти і визначення її рівня (нормальний - 20-30 мг на добу, підвищений - 3-8 г). Зразок сечі досліджується методом газової хроматографії або мас-спектрометрії, з використанням рідинної хроматографії; можливий скринінговий аналіз на наявність хлориду заліза в сечі. [14]
Також існує метод швидкої діагностики - визначення алкаптона в плямах висохлої сечі на папері (по інтенсивності кольору).
При уточненні діагнозу інструментальна діагностика (рентгенографія) передбачає виявлення у пацієнтів рентгенологічних ознак остеоартриту та інших суглобових патологій.
Підтверджують діагноз такі молекулярно-генетичні методи діагностика спадкових захворювань , як генетичне тестування і секвенування ДНК. [15]
Диференціальна діагностика
Диференціальна діагностика включає гемохроматоз і гостру печінкову недостатність новонароджених, меланінурію, гостру переміжну Порфирія, гемофагоцитарний лімфогістіоцитоз, первинну мітохондріальну патологію, ревматоїдний артрит, анкілозуючий спондиліт.
До кого звернутись?
Лікування алкаптонуріі
Основне лікування алкаптонуріі полягає в прийомі всередину великих доз (в день не менше 1000 мг) аскорбінової кислоти. У дітей це підвищує виведення гомогентизиновой кислоти з сечею, а у дорослих знижує рівень вмісту в сечі її дериватів - бензохінонуксусной кислоти - і уповільнює її зв'язування з сполучнотканинними структурами суглобів і колагеном. [16]
У західноєвропейських клініках проводять випробування ліків Нітізінон (Орфалін) - препарату групи метаболіка, який пригнічує другий етап катаболізму тирозину: трансформацію пара-гідроксіфенілпірувата в гомогентизинової кислоту. Однак застосування даного фармакологічного засоби призводить до накопичення тирозину і може викликати важкі побічні ефекти, включаючи помутніння рогівки і світлобоязнь, носові і шлункові кровотечі, недостатність печінки, зміни з боку крові та ін. Проте, в США Нітізінон схвалений FDA для лікування тірозінеміі I типу. [17], [18]
Як таке, фізіотерапевтичне лікування - ЛФК для збільшення м'язової сили і підвищення рухливості суглобів, бальнео і пелоідоттерапія для обмеження болю - проводиться при обумовлених Алкаптонурія проблеми з суглобами.
Хоча тирозин не тільки надходить з їжею, а й виробляється в організмі, пацієнтам з Алкаптонурія рекомендована дієта з низьким вмістом білка і обмеженням споживання продуктів, багатих тирозином, в першу чергу, яловичини і свинини, молочних продуктів (особливо сирів), бобових, горіхів і насіння.
Профілактика
Профілактика генних мутацій неможлива, а для попередження появи на світ дітей з високим ризиком вроджених порушень існує медико-генетичне консультування, яке необхідно перед запланованою вагітністю тих подружніх пар, в чиєму сімейному анамнезі є захворювання спадкового характеру. [19]
Прогноз
Випадків фатального результату перебігу алкаптонуріі дуже мало, і смерть може бути викликана серйозними ускладненнями, що зачіпають серце і нирки. Так що, для загальної тривалості життя людей з Алкаптонурія прогноз сприятливий.
Але якість життя знижується через інтенсивні болю в суглобах або хребті зі значним обмеженням рухливості, часто прогресуючим.