Медичний експерт статті

Нові публікації
Спадковий нефрит (синдром Альпорта) у дітей
Останній перегляд: 05.07.2025

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
Спадковий нефрит (синдром Альпорта) – генетично зумовлена спадкова неімунні гломерулопатія, що проявляється гематурією (іноді з протеїнурією), прогресуючим зниженням функції нирок з розвитком хронічної ниркової недостатності, часто поєднується з сенсоневральною глухотою та порушенням зору.
Вперше захворювання було описано в 1902 році Л. Г. Гатрі, який спостерігав сім'ю, в якій гематурія спостерігалася в кількох поколіннях. У 1915 році А. Ф. Херст описав розвиток уремії у членів цієї ж сім'ї. У 1927 році А. Альпорт вперше виявив втрату слуху у кількох родичів з гематурією. У 1950-х роках були описані ураження очей при подібному захворюванні. У 1972 році у пацієнтів зі спадковою гематурією під час морфологічного дослідження ниркової тканини Хінглейс та ін. виявили нерівномірне розширення та розшарування базальних мембран клубочків. У 1985 році була виявлена генетична основа спадкового нефриту - мутація в гені колагену IV типу (Fiengold et al., 1985).
Вивчення генетичної природи захворювання дозволило зробити висновок, що відмінності у фенотипових проявах спадкового нефриту (з втратою слуху або без неї) зумовлені ступенем експресії мутантного гена. Таким чином, наразі всі клінічні варіанти розглядаються як прояви одного захворювання, а термін «спадковий нефрит» є синонімом терміна «синдром Альпорта».
Згідно з епідеміологічними дослідженнями, спадковий нефрит зустрічається з частотою 17 на 100 000 дітей.
 [
[ Причини синдрому Альпорта
Генетичною основою захворювання є мутація в гені ланцюга а-5 колагену IV типу. Цей тип є універсальним для базальних мембран нирки, кохлеарного апарату, капсули кришталика, сітківки та рогівки ока, що було доведено в дослідженнях з використанням моноклональних антитіл проти цієї фракції колагену. Нещодавно вказана можливість використання ДНК-зондів для пренатальної діагностики спадкового нефриту.
Підкреслюється важливість тестування всіх членів сім'ї за допомогою ДНК-зондів для виявлення носіїв мутантного гена, що має велике значення при проведенні медико-генетичного консультування сімей з цим захворюванням. Однак до 20% сімей не мають родичів, які страждають на захворювання нирок, що свідчить про високу частоту спонтанних мутацій аномального гена. Більшість пацієнтів зі спадковим нефритом мають у своїх сім'ях осіб із захворюваннями нирок, втратою слуху та патологією зору; важливими є кровні шлюби між людьми з одним або кількома предками, оскільки у шлюбі споріднених осіб зростає ймовірність отримання однакових генів від обох батьків. Встановлено аутосомно-домінантний, аутосомно-рецесивний та домінантний, зчеплений з Х-хромосомою шляхи передачі.
У дітей найчастіше розрізняють три типи спадкового нефриту: синдром Альпорта, спадковий нефрит без втрати слуху та сімейна доброякісна гематурія.
Синдром Альпорта – спадковий нефрит із порушенням слуху. В основі його лежить комбінований дефект структури колагену клубочкової базальної мембрани нирок, структур вуха та ока. Ген класичного синдрому Альпорта розташований у локусі 21-22 q довгого плеча Х-хромосоми. У більшості випадків він успадковується домінантним чином, зчеплений з Х-хромосомою. У зв'язку з цим синдром Альпорта протікає важче у чоловіків, оскільки у жінок функція мутантного гена компенсується здоровим алелем другої, непошкодженої хромосоми.
Генетичною основою розвитку спадкового нефриту є мутації в генах альфа-ланцюгів колагену IV типу. Відомо шість альфа-ланцюгів колагену IV типу G: гени a5- та a6-ланцюгів (Col4A5 та Col4A5) розташовані на довгому плечі Х-хромосоми в зоні 21-22q; гени a3- та a4-ланцюгів (Col4A3 та Col4A4) – на 2-й хромосомі; гени a1- та a2-ланцюгів (Col4A1 та Col4A2) – на 13-й хромосомі.
У більшості випадків (80-85%) виявляється Х-зчеплений тип успадкування захворювання, пов'язаний з пошкодженням гена Col4A5 в результаті делеції, точкових мутацій або порушень сплайсингу. Наразі виявлено понад 200 мутацій гена Col4A5, відповідальних за порушення синтезу α5-ланцюгів колагену IV типу. При цьому типі успадкування захворювання проявляється у дітей обох статей, але у хлопчиків воно протікає важче.
Мутації в локусах генів Col4A3 та Col4A4, що відповідають за синтез ланцюгів a3 та a4 колагену IV типу, успадковуються аутосомно. Згідно з дослідженнями, аутосомно-домінантний тип успадкування спостерігається у 16% випадків спадкового нефриту, а аутосомно-рецесивний тип — у 6% пацієнтів. Відомо близько 10 варіантів мутацій генів Col4A3 та Col4A4.
Результатом мутацій є порушення процесів складання колагену IV типу, що призводить до порушення його структури. Колаген IV типу є одним з основних компонентів клубочкової базальної мембрани, кохлеарного апарату та кришталика ока, патологія якого буде виявлена в клініці спадкового нефриту.
Колаген IV типу, що входить до складу базальної мембрани клубочків, складається переважно з двох α1-ланцюгів (IV) та одного α2-ланцюга (IV), а також містить α3, α4, α5-ланцюги. Найчастіше при Х-зчепленому успадкуванні мутація гена Col4A5 супроводжується відсутністю α3-, α4-, α5- та α6-ланцюгів у структурі колагену IV типу, а кількість α1- та α2-ланцюгів у базальній мембрані клубочків збільшується. Механізм цього явища незрозумілий, передбачається, що причиною є посттранскрипційні зміни мРНК.
Відсутність ланцюгів a3, a4 та a5 у структурі колагену IV типу базальних мембран клубочків призводить до їх витончення та крихкості на ранніх стадіях синдрому Альпорта, що клінічно проявляється частіше гематурією (рідше гематурією з протеїнурією або лише протеїнурією), втратою слуху та лентиконусом. Подальше прогресування захворювання призводить до потовщення та порушення проникності базальних мембран на пізніх стадіях захворювання з проліферацією в них колагену V та VI типів, що проявляється у збільшенні протеїнурії та зниженні функції нирок.
Характер мутації, що лежить в основі спадкового нефриту, значною мірою визначає його фенотипічний прояв. У разі делеції Х-хромосоми з одночасною мутацією генів Col4A5 та Col4A6, відповідальних за синтез α5- та α6-ланцюгів колагену IV типу, синдром Альпорта поєднується з лейоміоматозом стравоходу та статевих органів. Згідно з даними досліджень, у разі мутації гена Col4A5, пов'язаної з делецією, відзначається більша тяжкість патологічного процесу, поєднання ураження нирок з позанирковими проявами та ранній розвиток хронічної ниркової недостатності, порівняно з точковою мутацією цього гена.
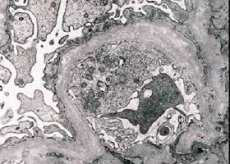
Морфологічно електронна мікроскопія виявляє витончення та розшарування базальних мембран клубочків (особливо lamina densa) та наявність електронно-щільних гранул. Ураження клубочків можуть бути гетерогенними у одного пацієнта, від мінімальних вогнищевих мезангіальних уражень до гломерулосклерозу. Гломеруліт при синдромі Альпорта завжди імунонегативний, що відрізняє його від гломерулонефриту. Характерними ознаками є розвиток канальцевої атрофії, лімфогістіоцитарна інфільтрація та наявність «пінистих клітин» з ліпідними включеннями – ліпофагів. У міру прогресування захворювання виявляються потовщення та виражене руйнування базальних мембран клубочків.
Виявляються певні зміни в імунній системі. У пацієнтів зі спадковим нефритом спостерігається знижений рівень Ig A та тенденція до підвищення концентрації IgM у крові, рівень IgG може бути підвищеним на ранніх стадіях захворювання та знижуватися на пізніх. Можливо, підвищення концентрації IgM та G є своєрідною компенсаторною реакцією у відповідь на дефіцит IgA.
Функціональна активність Т-лімфоцитарної системи знижена; відзначається селективне зменшення В-лімфоцитів, відповідальних за синтез Ig A, порушується фагоцитарна ланка імунітету, переважно через порушення хемотаксису та внутрішньоклітинних процесів травлення в нейтрофілах.
При дослідженні біопсії нирки у пацієнтів із синдромом Альпорта дані електронної мікроскопії виявляють ультраструктурні зміни клубочкової базальної мембрани: витончення, порушення структури та розщеплення клубочкової базальної мембрани зі зміною її товщини та нерівномірними контурами. На ранніх стадіях спадкового нефриту дефект визначає витончення та крихкість клубочкових базальних мембран.
Витончення клубочкових мембран є більш сприятливою ознакою і частіше зустрічається у дівчаток. Більш постійною електронно-мікроскопічною ознакою при спадковому нефриті є розщеплення базальної мембрани, причому тяжкість її руйнування корелює з тяжкістю процесу.
Симптоми синдрому Альпорта у дітей
Перші симптоми синдрому Альпорта у вигляді ізольованого сечового синдрому найчастіше виявляються у дітей перших трьох років життя. У більшості випадків захворювання виявляється випадково. Сечовий синдром виявляється під час профілактичного огляду дитини, перед надходженням до дитячого закладу або під час ГРВІ. У разі виявлення патології в сечі під час ГРВІ. При спадковому нефриті, на відміну від набутого гломерулонефриту, латентний період відсутній.
На початковій стадії захворювання здоров'я дитини мало страждає, характерною рисою є персистенція та стійкість сечового синдрому. Однією з основних ознак є гематурія різного ступеня тяжкості, що спостерігається у 100% випадків. Збільшення ступеня гематурії відзначається під час або після респіраторних інфекцій, фізичної активності або після профілактичних щеплень. Протеїнурія в більшості випадків не перевищує 1 г/добу, на початку захворювання може бути непостійною, у міру прогресування процесу протеїнурія посилюється. Періодично в сечовому осаді може бути присутня лейкоцитурія з переважанням лімфоцитів, що пов'язано з розвитком інтерстиціальних змін.
Згодом частково порушується функція нирок, погіршується загальний стан пацієнта: з'являються інтоксикація, м'язова слабкість, артеріальна гіпотензія, часто з'являються порушення слуху (особливо у хлопчиків), а іноді й порушення зору. Інтоксикація проявляється блідістю, втомою, головними болями. У початковій стадії захворювання втрата слуху в більшості випадків виявляється лише за допомогою аудіографії. Втрата слуху при синдромі Альпорта може виникати в різні періоди дитинства, але найчастіше втрата слуху діагностується у віці 6-10 років. Втрата слуху у дітей починається з високих частот, досягаючи значного ступеня в повітряній та кістковій провідності, переходячи від звукопровідної до звукосприймальної втрати слуху. Втрата слуху може бути одним з перших симптомів захворювання та може передувати сечовому синдрому.
У 20% випадків у пацієнтів із синдромом Альпорта спостерігаються зміни в органах зору. Найчастіше виявляються аномалії кришталика: сферофокія, передній, задній або змішаний лентиконус та різні катаракти. У сім'ях із синдромом Альпорта спостерігається значна частота міопії. Ряд дослідників постійно відзначають двосторонні перимакулярні зміни в цих сім'ях у вигляді яскравих білуватих або жовтуватих грануляцій у жовтому тілі. Вони вважають цю ознаку постійним симптомом, що має високу діагностичну цінність при синдромі Альпорта. К. С. Чуг та ін. (1993) в офтальмологічному дослідженні виявили у пацієнтів із синдромом Альпорта зниження гостроти зору у 66,7% випадків, передній лентиконус – у 37,8%, плями на сітківці – у 22,2%, катаракту – у 20%, кератоконус – у 6,7%.
У деяких дітей із спадковим нефритом, особливо при розвитку ниркової недостатності, відзначається значне відставання у фізичному розвитку. У міру прогресування ниркової недостатності розвивається артеріальна гіпертензія. У дітей вона частіше виявляється в підлітковому віці та у старших вікових групах.
Для пацієнтів зі спадковим нефритом характерна наявність різноманітних (більше 5-7) стигм дисморфогенезу сполучної тканини. Серед стигм сполучної тканини у пацієнтів найпоширенішими є гіпертелоризм очей, високе піднебіння, аномалії прикусу, аномальна форма вушних раковин, викривлення мізинця на руках та «сандальча щілина» на стопах. Спадковий нефрит характеризується одноманітністю стигм дисморфогенезу в межах сім'ї, а також високою частотою їх поширення серед родичів пробандів, по лінії яких передається захворювання.
На ранніх стадіях захворювання виявляється ізольоване зниження часткових функцій нирок: транспорту амінокислот, електролітів, концентраційної функції, ацидогенезу, пізніші зміни впливають на функціональний стан як проксимальної, так і дистальної частин нефрона та характеризуються комбінованими частковими порушеннями. Зниження клубочкової фільтрації відбувається пізніше, частіше в підлітковому віці. У міру прогресування спадкового нефриту розвивається анемія.
Таким чином, спадковий нефрит характеризується стадійним перебігом захворювання: спочатку латентна стадія або прихована клінічна симптоматика, що проявляється мінімальними змінами сечового синдрому, потім настає поступова декомпенсація процесу зі зниженням функції нирок з маніфестними клінічними симптомами (інтоксикація, астенія, затримка розвитку, анемія). Клінічні симптоми зазвичай з'являються незалежно від нашарування запальної реакції.
Спадковий нефрит може проявлятися в різні вікові періоди, що залежить від дії гена, який до певного часу знаходиться в репресованому стані.
Класифікація
Існує три типи спадкового нефриту
- I варіант – клінічно проявляється як нефрит з гематурією, втратою слуху та ураженням очей. Перебіг нефриту прогресуючий з розвитком хронічної ниркової недостатності. Тип успадкування – домінантний, зчеплений з Х-хромосомою. Морфологічно виявляється порушення структури базальної мембрани, її витончення та розщеплення.
- II варіант – клінічно проявляється як нефрит з гематурією без втрати слуху. Перебіг нефриту прогресуючий з розвитком хронічної ниркової недостатності. Тип успадкування домінантний, зчеплений з Х-хромосомою. Морфологічно виявляється витончення базальної мембрани клубочкових капілярів (особливо laminadensa).
- III варіант – доброякісна сімейна гематурія. Перебіг сприятливий, хронічна ниркова недостатність не розвивається. Тип успадкування – аутосомно-домінантний або аутосомно-рецесивний. При аутосомно-рецесивному типі успадкування у жінок відзначається важчий перебіг захворювання.
Діагностика синдрому Альпорта
Пропонуються такі критерії:
- наявність щонайменше двох пацієнтів з нефропатією в кожній родині;
- гематурія як провідний симптом нефропатії у пробанда;
- наявність втрати слуху хоча б у одного члена сім'ї;
- розвиток хронічної ниркової недостатності у одного або кількох родичів.
У діагностиці різних спадкових та вроджених захворювань велике місце відводиться комплексному підходу до обстеження та, перш за все, звертанню уваги на дані, отримані під час складання родоводу дитини. Діагноз синдрому Альпорта вважається валідним у випадках, коли у пацієнта виявлено 3 з 4 типових ознак: наявність гематурії та хронічної ниркової недостатності в родині, наявність нейросенсорної втрати слуху, патології зору у пацієнта, виявлення ознак розщеплення базальної мембрани клубочків зі зміною її товщини та нерівними контурами під час електронно-мікроскопічної характеристики біопсії.
Обстеження пацієнта повинно включати клініко-генетичні методи дослідження; цілеспрямоване вивчення анамнезу захворювання; загальний огляд пацієнта з урахуванням діагностично значущих критеріїв. У стадії компенсації патологію можна виявити, лише зосередившись на таких синдромах, як наявність спадкового обтяженості, гіпотензія, множинні стигми дизембріогенезу, зміни сечового синдрому. У стадії декомпенсації можуть з'являтися позаниркові симптоми, такі як виражена інтоксикація, астенія, затримка фізичного розвитку, анемія, що проявляються та посилюються з поступовим зниженням функції нирок. У більшості пацієнтів при зниженні функції нирок спостерігається: зниження ацидо- та аміногенезу; 50% пацієнтів відзначають значне зниження секреторної функції нирок; обмежений діапазон коливань оптичної щільності сечі; порушення ритму фільтрації, а потім зниження клубочкової фільтрації. Стадія хронічної ниркової недостатності діагностується, коли у пацієнтів спостерігається підвищений рівень сечовини в сироватці крові (більше 0,35 г/л) протягом 3-6 місяців і більше, а також зниження клубочкової фільтрації до 25% від норми.
Диференціальну діагностику спадкового нефриту слід проводити насамперед з гематуричною формою набутого гломерулонефриту. Набутий гломерулонефрит найчастіше має гострий початок, період 2-3 тижнів після інфекції, позаниркові ознаки, включаючи гіпертензію з перших днів (при спадковому нефриті, навпаки, гіпотензія), зниження клубочкової фільтрації на початку захворювання, відсутність порушень часткових канальцевих функцій, тоді як при спадковому вони присутні. Набутий гломерулонефрит протікає з більш вираженою гематурією та протеїнурією, зі збільшенням ШОЕ. Типові зміни базальної мембрани клубочків, характерні для спадкового нефриту, мають діагностичне значення.
Диференціальна діагностика з дисметаболічною нефропатією проводиться з хронічною нирковою недостатністю, в сім'ї клінічно виявлені гетерогенні захворювання нирок, і може спостерігатися спектр нефропатії від пієлонефриту до сечокам'яної хвороби. Діти часто скаржаться на біль у животі та періодично під час сечовипускання, в осаді сечі - оксалати.
Якщо є підозра на спадковий нефрит, пацієнта слід направити до спеціалізованого нефрологічного відділення для уточнення діагнозу.
Що потрібно обстежити?
Як обстежувати?
Які аналізи необхідні?
До кого звернутись?
Лікування синдрому Альпорта
Режим включає обмеження важких фізичних навантажень та перебування на свіжому повітрі. Дієта повноцінна, з достатнім рівнем повноцінних білків, жирів та вуглеводів, з урахуванням функції нирок. Велике значення має виявлення та лікування хронічних вогнищ інфекції. Використовуються такі медикаменти: АТФ, кокарбоксилаза, піридоксин (до 50 мг/добу), карнітин хлорид. Курси проводяться 2-3 рази на рік. При гематурії призначають фітотерапію – кропиву дводомну, сік аронії, деревій.
У зарубіжній та вітчизняній літературі є повідомлення про лікування преднізолоном та застосування цитостатиків. Однак важко судити про ефект.
При хронічній нирковій недостатності застосовують гемодіаліз та трансплантацію нирки.
Методів специфічної (ефективної патогенетичної) терапії спадкового нефриту не існує. Усі лікувальні заходи спрямовані на запобігання та уповільнення зниження функції нирок.
Раціон має бути збалансованим та висококалорійним, враховуючи функціональний стан нирок. За відсутності функціональних порушень раціон дитини повинен містити достатню кількість білків, жирів та вуглеводів. За наявності ознак порушення функції нирок кількість білків, вуглеводів, кальцію та фосфору слід обмежувати, що затримує розвиток хронічної ниркової недостатності.
Фізичну активність слід обмежити; дітям рекомендується уникати занять спортом.
Слід уникати контактів з інфекційними хворими, знизити ризик розвитку гострих респіраторних захворювань. Необхідна санація вогнищ хронічної інфекції. Дітям із спадковим нефритом профілактичні щеплення не проводяться, вакцинація можлива лише за епідеміологічними показаннями.
Гормональна та імуносупресивна терапія при спадковому нефриті неефективна. Є вказівки на деякий позитивний ефект (зменшення протеїнурії та уповільнення прогресування захворювання) при тривалому багаторічному застосуванні циклоспорину А та інгібіторів АПФ.
У лікуванні пацієнтів використовуються препарати, що покращують обмін речовин:
- піридоксин – 2-3 мг/кг/добу в 3 прийоми протягом 4 тижнів;
- кокарбоксилаза – 50 мг внутрішньом’язово через день, всього 10-15 ін’єкцій;
- АТФ – 1 мл внутрішньом’язово через день, 10-15 ін’єкцій;
- вітамін А – 1000 МО/рік/день в 1 прийом протягом 2 тижнів;
- Вітамін Е – 1 мг/кг/день в 1 прийом протягом 2 тижнів.
Цей вид терапії допомагає покращити загальний стан пацієнтів, зменшити порушення функції канальців і проводиться курсами 3 рази на рік.
Левамізол можна використовувати як імуномодулятор – 2 мг/кг/добу 2-3 рази на тиждень з перервами між прийомами 3-4 дні.
Згідно з даними досліджень, гіпербарична оксигенація позитивно впливає на тяжкість гематурії та порушення функції нирок.
Найефективнішим методом лікування спадкового нефриту є своєчасна трансплантація нирки. У цьому випадку рецидиву захворювання в трансплантованій нирці не відбувається; у невеликому відсотку випадків (близько 5%) нефрит може розвинутися в трансплантованій нирці, пов'язаний з антигенами до базальної мембрани клубочків.
Перспективним напрямком є пренатальна діагностика та генно-інженерна терапія. Експерименти на тваринах показують високу ефективність перенесення нормальних генів, відповідальних за синтез альфа-ланцюгів колагену IV типу, у тканину нирок, після чого спостерігається синтез нормальних колагенових структур.
Прогноз
Прогноз при спадковому нефриті завжди серйозний.
Прогностично несприятливими критеріями перебігу спадкового нефриту є:
- чоловіча стать;
- ранній розвиток хронічної ниркової недостатності у членів сім'ї;
- протеїнурія (більше 1 г/добу);
- потовщення базальних мембран клубочків за даними мікроскопії;
- акустичний неврит;
- делеція в гені Col4A5.
Прогноз при доброякісній сімейній гематурії більш сприятливий.
Использованная литература