Медичний експерт статті

Нові публікації
Хвороба Шарко-Марі-Тута
Останній перегляд: 12.07.2025

Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
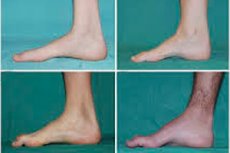
Перонеальна м'язова атрофія, синдром або хвороба Шарко-Марі-Тута – це група хронічних спадкових захворювань з ураженням периферичних нервів.
Згідно з МКХ-10, у розділі про хвороби нервової системи код цього захворювання – G60.0 (спадкова рухова та сенсорна нейропатія). Воно також входить до переліку орфанних захворювань.
Епідеміологія
Згідно з клінічною статистикою, поширеність усіх видів хвороби Шарко-Марі-Тута на 100 тисяч населення становить 19 випадків (за іншими даними, один випадок на 2,5-10 тисяч населення).
На карциному мієломи 1 типу припадає близько двох третин випадків (один випадок на 5000–7000 населення), і майже 70% з них пов’язані з дуплікацією гена PMP22. Понад 1,2 мільйона людей у світі страждають від цього типу захворювання.
Захворюваність на ХМТ 4 типу оцінюється в 1-5 випадків на 10 000 дітей. [ 1 ]
Причини хвороби Шарко-Марі-Тута
Згідно з класифікацією полінейропатичних синдромів, перонеальна (фібулярна) м'язова атрофія, нейронна аміотрофія Шарко-Марі-Тута або хвороба Шарко-Марі-Тута (скорочено ХШТ) належить до генетично зумовлених моторно-сенсорних полінейропатій. [ 2 ]
Тобто, причинами його виникнення є генетичні мутації. І залежно від характеру генетичних відхилень розрізняють основні типи або види цього синдрому: демієлінізуючий та аксональний. До першої групи належить хвороба Шарко-Марі-Тута 1 типу (ХШМТ1), яка виникає внаслідок дуплікації гена PMP22 на 17-й хромосомі, що кодує трансмембранний периферичний мієліновий білок 22. В результаті відбувається сегментарна демієлінізація оболонки аксона (відростків нервових клітин) та зниження швидкості проведення нервового сигналу. Крім того, мутації можуть бути в деяких інших генах.
Аксональна форма — це хвороба Шарко-Марі-Тута 2 типу (ХШМТ2), яка вражає самі аксони та пов'язана з патологічними змінами в гені MFN2 у локусі 1p36.22, що кодує мембранний білок мітофузин-2, необхідний для злиття мітохондрій та формування функціональних мітохондріальних мереж всередині периферичних нервових клітин. Існує понад десяток підтипів ХШМТ2 (з мутаціями в певних генах).
Слід зазначити, що наразі виявлено понад сто генів, пошкодження яких, що передаються у спадок, спричиняють різні підтипи хвороби Шарко-Марі-Тута. Наприклад, мутації в гені RAB7 призводять до ХМТ типу 2B; зміна гена SH3TC2 (що кодує один з мембранних білків клітин Шванна) спричиняє ХМТ типу 4C, яка проявляється в дитячому віці та характеризується демієлінізацією рухових та сенсорних нейронів (існує півтора десятка форм 4 типу цієї хвороби).
Рідкісний CMT 3 типу (так званий синдром Дежеріна-Соттаса) починає розвиватися в ранньому дитинстві та спричинений мутаціями в генах PMP22, MPZ, EGR2 та інших.
При виникненні ШМТ 5 типу у віці 5-12 років спостерігається не тільки рухова нейропатія (у вигляді спастичного парапарезу нижніх кінцівок), але й ураження зорового та слухового нервів.
М'язова слабкість та атрофія зорового нерва (з втратою зору), а також проблеми з рівновагою є типовими для ХМТ 6 типу. А при хворобі Шарко-Марі-Тута 7 типу спостерігається не лише моторно-сенсорна нейропатія, але й захворювання сітківки у вигляді пігментного ретиніту.
Х-зчеплена Х-хромосома ХМТ або хвороба Шарко-Марі-Тута з тетрапарезом кінцівок (слабкість рухів обох рук і ніг) частіше зустрічається у чоловіків, є демієлінізуючим типом і вважається наслідком мутації в гені GJB1 на довгому плечі Х-хромосоми, який кодує коннексин 32, трансмембранний білок клітин Шванна та олігодендроцитів, що регулює передачу нервових сигналів. [ 3 ]
Фактори ризику
Основним фактором ризику розвитку ХМТ є сімейний анамнез захворювання, тобто у близьких родичів.
За даними генетиків, якщо обоє батьків є носіями аутосомно-рецесивного гена хвороби Шарко-Марі-Тута, ризик народження дитини, у якої розвинеться це захворювання, становить 25%. А ризик того, що дитина буде носієм цього гена (але не матиме жодних симптомів), оцінюється в 50%.
У випадку Х-зчепленого успадкування (коли мутований ген знаходиться на Х-хромосомі жінки) існує 50% ризик того, що мати передасть ген своєму синові, у якого розвинеться ХМТ. Захворювання може не виникнути при народженні дівчинки, але сини (онуки) доньки можуть успадкувати дефектний ген, і захворювання розвинеться.
Патогенез
При будь-якому типі хвороби Шарко-Марі-Тута її патогенез зумовлений спадковою аномалією периферичних нервів: рухових (рухових) та сенсорних (чутливих).
Якщо тип CMT є демієлінізуючим, то руйнування або дефект мієлінової оболонки, що захищає аксони периферичних нервів, призводить до уповільнення передачі нервових імпульсів у периферичній нервовій системі – між мозком, м’язами та органами чуття.
При аксональному типі захворювання уражаються самі аксони, що негативно впливає на силу нервових сигналів, якої недостатньо для повноцінної стимуляції м’язів та органів чуття.
Читайте також:
Як передається синдром Шарко-Марі-Тута? Дефектні гени можуть успадковуватися аутосомно-домінантним або аутосомно-рецесивним типом.
Найпоширеніший тип, аутосомно-домінантне успадкування, виникає, коли є одна копія мутованого гена (носія одного з батьків). А ймовірність передачі CMT кожній народженій дитині оцінюється в 50%. [ 4 ]
При аутосомно-рецесивному успадкуванні для розвитку захворювання необхідні дві копії дефектного гена (по одній від кожного з батьків, які не проявляють жодних ознак захворювання).
У 40-50% випадків відбувається аутосомно-домінантна спадкова демієлінізація, тобто CMT 1 типу; у 12-26% випадків - аксональна CMT, тобто 2 типу. А в 10-15% випадків спостерігається Х-зчеплене успадкування. [ 5 ]
Симптоми хвороби Шарко-Марі-Тута
Зазвичай перші ознаки цього захворювання починають проявлятися в дитинстві та підлітковому віці та поступово розвиваються протягом життя, хоча синдром може дати про себе знати пізніше. Поєднання симптомів є мінливим, і швидкість прогресування захворювання, як і його тяжкість, неможливо передбачити.
Типовими симптомами початкової стадії є підвищена загальна стомлюваність; знижений тонус (слабкість) м'язів стоп, щиколоток та гомілок; відсутність рефлексів. Це ускладнює рух стоп і призводить до дисбазії (порушення ходи) у вигляді більш високого підйому ніг, часто з частими спіткненнями та падіннями. Ознаками хвороби Шарко-Марі-Тута у маленької дитини можуть бути виражена незграбність та нехарактерні для віку труднощі з ходьбою, пов'язані з двостороннім опущенням стопи. Характерні також деформації стоп: високий склепіння (запавшина стопа) або виражене плоскостопість, викривлені (молоткоподібні) пальці.
У разі ходьби на носках на тлі м’язової гіпотонії невролог може запідозрити у дитини ХМТ 4 типу, при якій діти можуть бути не в змозі ходити до підліткового віку.
У міру прогресування захворювання м’язова атрофія та слабкість поширюються на верхні кінцівки, що ускладнює виконання дрібної моторики та звичайних завдань руками. Зниження тактильних відчуттів та здатності відчувати тепло та холод, а також оніміння в стопах і руках свідчать про пошкодження аксонів сенсорних нервів.
При хворобі Шарко-Марі-Тута 3 та 6 типів, що проявляється в дитячому віці, спостерігаються сенсорна атаксія (порушення координації рухів та рівноваги), посмикування м'язів та тремор, пошкодження лицевого нерва, атрофія зорового нерва з ністагмом та втрата слуху.
На пізніх стадіях може спостерігатися неконтрольоване тремтіння (тремор) та часті м’язові судоми; проблеми з рухом можуть призвести до розвитку болю: м’язового, суглобового, невропатичного.
Ускладнення і наслідки
Хвороба Шарко-Марі-Тута може мати такі ускладнення та наслідки, як:
- частіші розтягнення зв'язок та переломи;
- контрактури, пов'язані зі скороченням навколосуглобових м'язів і сухожиль;
- сколіоз (викривлення хребта);
- проблеми з диханням – коли пошкоджені нервові волокна, що іннервують м’язи діафрагми:
- втрата здатності самостійно пересуватися.
Діагностика хвороби Шарко-Марі-Тута
Діагностика включає клінічний огляд, анамнез (включаючи сімейний анамнез), неврологічне та системне обстеження.
Проводяться тести для перевірки діапазону рухів, чутливості та сухожильних рефлексів. Нервову провідність можна оцінити за допомогою інструментальної діагностики – електроміографії або електронейроміографії. Також може знадобитися ультразвукове дослідження або МРТ. [ 6 ]
Генетичне або ДНК-тестування для виявлення найпоширеніших генетичних мутацій, що викликають карциному-мезенхімальну міалгію (КМТ), у зразку крові обмежене, оскільки ДНК-тести наразі доступні не для всіх типів захворювання. Для отримання додаткової інформації див. Генетичне тестування.
У деяких випадках проводиться біопсія периферичного нерва (зазвичай литкового нерва).
Диференціальна діагностика
Диференціальна діагностика включає інші периферичні нейропатії, м'язову дистрофію Дюшена, мієлопатичний та міастенічний синдроми, діабетичну нейропатію, мієлопатії при розсіяному та аміотрофічному бічному склерозі, синдром Гійєна-Барре, травму малогомілкового нерва та його атрофію (в тому числі при защемленні між поперековими дисками хребта), пошкодження мозочка або таламуса, а також побічні ефекти хіміотерапії (під час лікування цитостатиками, такими як вінкристин або паклітаксел). [ 7 ]
До кого звернутись?
Лікування хвороби Шарко-Марі-Тута
Сьогодні лікування цього спадкового захворювання включає лікувальну фізкультуру (спрямовану на зміцнення та розтягування м’язів); трудотерапію (яка допомагає пацієнтам зі слабкістю м’язів у руках); та використання ортопедичних пристроїв для полегшення ходьби. За необхідності призначають знеболювальні або протисудомні препарати. [ 8 ]
У випадках тяжкого плоскостопості може бути проведена остеотомія, а у разі деформації п'яти показана хірургічна корекція – артродез. [ 9 ]
Тривають дослідження як генетичної складової захворювання, так і методів його лікування. Використання стовбурових клітин, деяких гормонів, лецитину чи аскорбінової кислоти поки що не дало позитивних результатів.
Але завдяки нещодавнім дослідженням, у найближчому майбутньому справді може з'явитися щось нове в лікуванні хвороби Шарко-Марі-Тута. Так, з 2014 року французька компанія Pharnext розробляє, а з середини 2019 року тривають клінічні випробування препарату PXT3003 для лікування ХМТ 1 типу у дорослих, який пригнічує підвищену експресію гена PMP22, покращує мієлінізацію периферичних нервів та полегшує нервово-м'язові симптоми.
Фахівці медичної компанії Sarepta Therapeutics (США) працюють над створенням генної терапії для лікування хвороби Шарко-Марі-Тута 1 типу. У цій терапії буде використано нешкідливий аденоасоційований вірус (AAV) роду Dependovirus з лінійним одноланцюговим ДНК-геномом, який перенесе в організм ген NTF3, що кодує білок нейротрофін-3 (NT-3), необхідний для функціонування нервових клітин Шванна.
Helixmith розпочне клінічні випробування розробленої в Південній Кореї генної терапії Engensis (VM202) до кінця 2020 року для лікування м’язових симптомів при карциномі мочеточника 1 типу. [ 10 ]
Профілактика
Профілактикою ХМТ може бути генетичне консультування майбутніх батьків, особливо якщо у когось із подружжя є сімейний анамнез захворювання. Однак виявлені випадки точкових генних мутацій de novo, тобто за відсутності захворювання в сімейному анамнезі.
Під час вагітності ймовірність хвороби Шарко-Марі-Тута у майбутньої дитини можна перевірити за допомогою біопсії ворсин хоріона (з 10 по 13 тиждень вагітності), а також аналізу навколоплідних вод (на 15-18 тижні).
Прогноз
Загалом, прогноз для різних типів хвороби Шарко-Марі-Тута залежить від клінічної тяжкості, але в усіх випадках захворювання прогресує повільно. Багато пацієнтів мають інвалідність, хоча це не скорочує тривалість життя.